Introduction
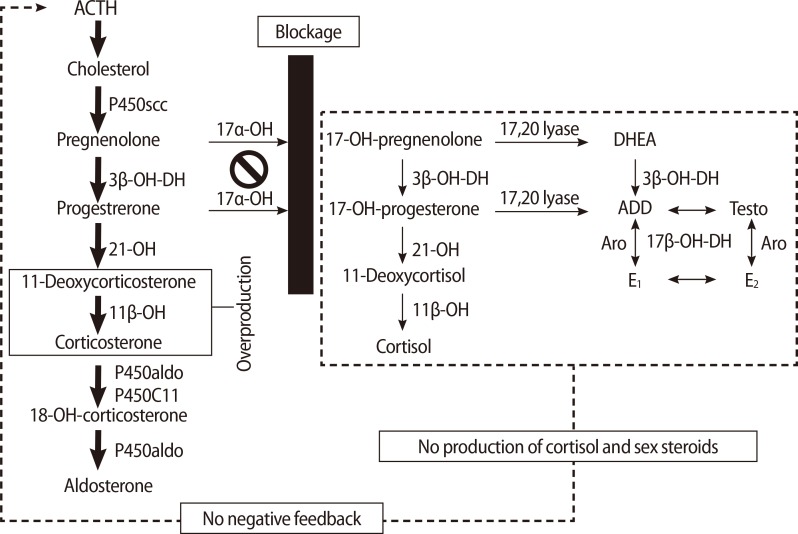
17α-hydroxylase and 17,20-lyase (P450c17) are enzymes that convert pregnenolone and progesterone to 17α-hydroxypregnenolone (17-OHPreg) and 17α-hydroxyprogesterone (17-OHP), which are precursors of sex steroids and cortisol. In cases of P450c17 deficiency, androgens, estrogens, and cortisol cannot be produced. A compensatory increase in adrenocorticotrophic hormone (ACTH), due to the failure of cortisol production, stimulates the production of 11-deoxycorticosterone and corticosterone, which have powerful mineralocorticoid activity. In turn, the excessive levels of these mineralocorticoids lead to volume expansion, hypertension, and electrolyte imbalances such as hypokalemia (Figure 1).
Deficiency in sex steroids and excessive mineralocorticoid production cause characteristic symptoms and signs such as delayed puberty with primary amenorrhea and hypertension. Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder due to a defect in any of several enzymes involved in steroidogenesis; different genotypes induce different phenotypes. 17α-hydroxylase/17,20-lyase deficiency (17OHD) is a rare type of CAH that causes cortisol and sex hormone deficiency and aldosterone excess [1]. Here, we report the case of a 21-year-old woman who had been inadequately treated for 17α-hydroxylase/17,20-lyase deficiency for a prolonged period of time.
Case report
An 18-year-old female was transferred for evaluation of low serum potassium levels (2.6 mmol/L) found incidentally during a preoperative workup for an appendectomy at a local medical center. The patient had no past medical or family history associated with electrolyte imbalance. The hypokalemia appeared to be corrected after potassium supplementation. Then, the patient was referred to the cardiology department because she had constant high blood pressure, with a maximal reading of 180/110 mm Hg. The patient was treated with an angiotension II receptor antagonist (telmisartan) and a calcium channel blocker (amlodipine). However, her blood pressure was still not well controlled and she was subsequently diagnosed with stage 1 hypertensive retinopathy.
Upon further investigation, it became apparent that the patient had had hypertension since the age of 15-year-old, when the patient visited her gynecologist. At that time, the patient was found to have primary amenorrhea and features of sexual infantilism. On physical examination, the patient was quite tall and slender, with a height of 166.2 cm and a body weight of 44 kg (body mass index, 15.9 kg/m2), and was phenotypically female. The patient's breast development had only progressed to Tanner stages 1-2 and pubic hair was underdeveloped. Ultrasonography of the abdomen and pelvis demonstrated a hypoplastic uterine corpus, bilateral small multicystic ovaries, and a very thin endometrium. Considering the patient's history and clinical signs, hormone profiles and karyotype were checked. The patient's karyotype was 46,XX and hormone studies showed a normal to hypergonadotropic hypogonadal state with low testosterone and dehydroepiandrosterone sulfate (DHEA-S) levels (Table 1). Because there was a high suspicion of 17OHD, further evaluation was recommended; however, the patient refused further investigation at this time. After a period of 4.5 years, the patient revisited the gynecology department with a complaint of persistent amenorrhea. Upon physical examination, there was no interval change in the patient's sexual development. A repeat hormone profile showed elevated levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) with low levels of estradiol, indicating hypergonadotropic hypogonadism. Studies also demonstrated very low cortisol, high ACTH, and low levels of both testosterone and DHEA-S (Table 1). With a clinical suspicion of an enzyme defect in steroidogenesis, particularly 17OHD, an ACTH stimulation test and a CYP17A1 gene analysis were performed. The ACTH stimulation test showed no increase in 17-OHP after intravenous administration of synthetic ACTH (Synacthen 0.25 mg, Dalim Biotech, Jecheon, Korea) when compared with the basal level (Table 2), suggesting an impairment in the pathway catalyzed by 17α-hydroxylase. Genetic analysis showed a compound heterozygous mutation in exon 6 of the CYP17A1 gene (c.985-987TAC>AA and c.1118A>T). After being diagnosed with 17α-hydroxylase deficiency, the patient stopped taking antihypertensive medication and started oral hydrocortisone (Rapison 30 mg daily, Kolon Pharmaceuticals, Daejeon, Korea), which is a cornerstone for the treatment for CAH. In addition, the patient commenced oral estradiol and cyclic medroxyprogesterone (Progynova 2 mg daily, Bayer, Lys-Lez-Lannoy, France and Provera 10 mg daily for 13 days each month, Pfizer, Tronto, Italy) in order to induce secondary sexual development and maintain cyclic uterine bleeding. After appropriate therapy, the patient's blood pressure normalized without antihypertensive therapy, and five months later, follow up laboratory investigations revealed normal levels of ACTH (11.04 µg/dL), progesterone (0.04 ng/mL) and serum electrolytes. Physical examination showed stage 2 Tanner breast development with scanty pubic and axillary hair production, and the uterus was normal in size and shape upon ultrasonographic examination.
Discussion
17α-hydroxylase deficiency is a rare form of CAH, with an estimated incidence of 1 in 50,000 to 100,000, and accounts for about 1% of all CAH cases [2,3]. The most common enzyme defect causing CAH is 21-hydroxylase deficiency (21-OHD), followed by 11β-hydroxylase deficiency (11β-OHD). Clinically, these enzyme deficiencies are characterized by hyperandrogenism or heterosexual precocious puberty in females; however, clinical features of 17α-hydroxylase deficiency differ from those of 21-OHD or 11β-OHD in that there is no hyperandrogenism, but rather, sexual infantilism.
The enzyme P450c17 has both 17α-hydroxylase and 17,20-lyase activity. The gene encoding the enzyme P450c17 is CYP17A1, which is located at chromosome 10q24.3. Human 17α-hydroxylase deficiency involving a loss of only 17α-hydroxylase or 17,20-lyase has been observed [4,5], but in most affected patients, both enzymes are deficient [6]. Blockage of the pathway catalyzed by 17α-hydroxylase enhances the mineralocorticoid pathway, resulting in overproduction of 11-DOC, corticosterone and aldosterone. This also causes a decline in 17-OHPreg, 17-OHP, 11-deoxycortisol, cortisol, DHEA, androstenedione, and testosterone [1]. High levels of 11-DOC induce sodium and fluid retention and loss of potassium and hydrogen, and consequently hypertension, because of its potent mineralocorticoid effect [7]. Affected males have ambiguous or female external genitalia due to a deficiency in androgen synthesis. Female patients have normal genitalia but show immature sexual development and primary amenorrhea caused by a deficiency of estrogen in adolescence [1]. Once diagnosis is made, immediate adequate intervention should be initiated in order to prevent complications such as hypertension or hypokalemia and to induce sexual development at the appropriate time.
The diagnosis of 17OHD is based on clinical, biochemical, and molecular features. In our case, biochemically, there were decreased concentrations of DHEA, androstenedione, testosterone, estradiol, and cortisol, and increased concentrations of 11-DOC and ACTH. Since clinical and biochemical features are diverse, depending on the type of mutations in the CYP17A1 gene, genetic analysis is critical for confirmative diagnosis [8]. Some genetic abnormalities such as point mutations by single base substitution, small duplications, and large deletions have been reported in 17OHD [9,10]. In our case, compound heterozygous mutations in exon 6 of the CYP17A1 gene (c.985-987TAC>AA and c.1118A>T) were detected. The substitution of the TAC codon at 329 with AA is assumed to induce a frameshift variation such that consequently, the codon at 418 becomes a stop codon; additionally, a missense mutation is developed via the substitution of histidine with leucine at codon 373. These mutations are some of the frequently reported mutations of the CYP17A1 gene in Asian countries such as Korea, Japan, and China [10]. Of the mutations reported among the Korean population, H373L mutation (c.1118A>T) was the most common; additional mutations are also reported (Table 3) [8,11]. Because 17OHD is an autosomal recessive disorder, we investigated the family history. While there were no affected individuals apparent from the clinical history, we recommended genetic analysis of the patient's parents; however, this was not performed due to a lack of consent.
Treatment of 17OHD comprises appropriate glucocorticoid and sex steroid hormone supplementation. Glucocorticoids are administered in order to decrease and normalize the blood levels of 11-DOC and ACTH. Glucocorticoid therapy can normalize blood pressure and serum electrolyte levels. Generally, dexamethasone (0.25-1.0 mg/day), prednisone (2-5 mg/day), or equivalents are recommended. Sex hormone replacement is required for breast and uterus development and maintenance of female sexual characteristics. Estrogen and progestin agents are required in patients with a 46,XX karyotype to induce cyclic withdrawal bleeding and to prevent endometrial hyperplasia.
Our patient had not been managed appropriately for a prolonged period prior to consultation with us. The patient had received intermittent potassium replacement and antihypertensive treatment following the incomplete diagnosis of her clinical condition. After administration of glucocorticoid (hydrocortisone 10 mg three times daily) and sex steroids (estradiol valerate 2 mg once daily and medroxyprogesterone acetate 10 mg once daily for 13 days per month), the patient's blood levels of renin, aldosterone, ACTH, and progesterone were normalized.
Patients with 17OHD cannot produce their own mature oocytes because of failed follicular development due to the irreversible defect in steroidogenesis, and therefore cannot conceive spontaneously or via endocrinologic intervention. Theoretically, assisted reproductive technology using in vitro maturation of immature oocytes is the only way for these female patients to have their own genetic offspring. Otherwise, an oocyte donation program is an alternative means of conception.
17OHD is a rare and peculiar endocrine disorder. Almost all patients present for the evaluation of delayed puberty and may not complain of symptoms until the time of puberty, thus an early diagnosis is difficult to make prior to this clinical phase. Some patients may visit the hospital at an earlier age for evaluation of hypertension that comes from increased 11-DOC before menarche. For early diagnosis and adequate management, 17OHD should be considered when a girl or a young woman presents with poor sexual development and hypertension, because hypertension is uncommon in young age.