Introduction
Insertions occur when a single chromosome breaks at 2 places and a piece of the chromosome is reinserted elsewhere. The piece of chromosome may be reinserted into a different location in the original chromosome (intrachromosomal insertion) or it may be reinserted into a different chromosome (interchromosomal insertion). Intrachromosomal insertion can occur on the same side of the centromere (paracentric) or on the opposite side of the centromere (pericentric).
Intrachromosomal insertions are very rare forms of chromosomal rearrangements; about 40 cases have been published [1-4]. Balanced carriers of intrachromosomal insertions are phenotypically normal, except a case report of cri-du-chat syndrome [5]. On the other hand, they are at risk for unbalanced meiotic segregation due to crossing over, and the frequency of a child having an unbalanced karyotype is about 15-50% [2].
Patients with trisomy 4q syndrome have variable clinical features, often including growth and developmental delays, mental retardation, and a dysmorphic appearance [6]. Pure partial trisomy of 4q may contribute significantly to the clarification of region specific phenotypes, but very few cases have been reported. In this study, we present an intrachromosomal insertion duplication of chromosome 4q that was detected in an infertile man with non-obstructive azoospermia.
Case report
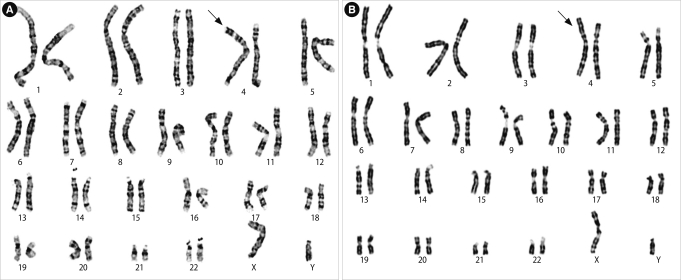
A 35-year-old man with infertility was referred for chromosomal analysis. His height was 175 cm and weight was 63 kg. The results of semen analysis according to the guidelines of the World Health Organization [7] showed azoospermia. The histological examination of the testicular tissue revealed spermatogenic arrest in meiosis, confirming the diagnosis of non-obstructive azoospermia. He was diagnosed with sertoli-cell-only syndrome because all the seminiferous tubules contained only Sertoli cells and did not contain germ cells. He was psychologically normal. Chromosome analysis was performed using Giemsa banding according to usual procedures. The patient was seen to have additional material of unknown origin on the terminal region of the short arm of chromosome 4 (4p). The additional material was relatively large in size with two dark bands.
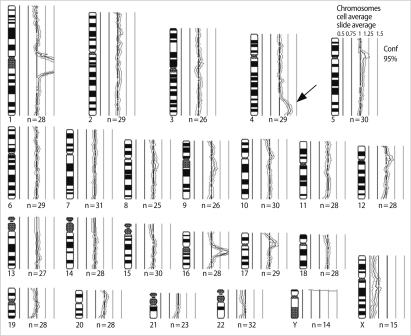
To confirm the origin of the unknown material, we carried out high-resolution banding, comparative genomic hybridization (CGH) and FISH. High-resolution GTG and RBG banding analysis showed an unbalanced karyotype with additional material attached to the terminal band of 4p16 (Figure 1). However, we could not estimate the origin of the extra segment or the type of rearrangement. DNA was extracted from the peripheral blood of the patient and used in a CGH method. CGH revealed a gain of material on the region of 4q32-35 (Figure 2). FISH was carried out with multiple probes for chromosome 4, including the whole chromosome painting (wcp) probe, telomeric probes for 4p (D4S3359)/4q (D4S2930), and the Wolf-Hirschhorn region (D4S96)/centromeric (D4Z1) probe (Vysis, Downers Grove, IL, USA).
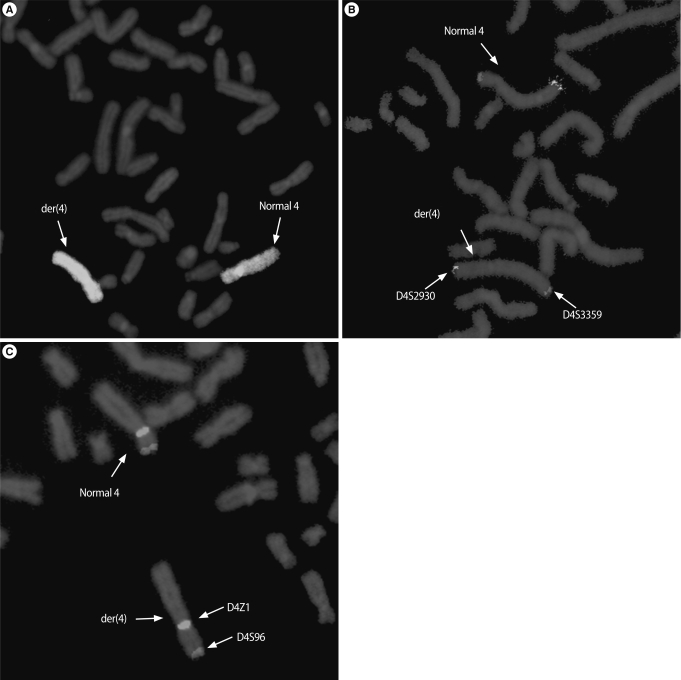
For each analysis, a minimum of ten metaphase and 100 interphase cells were scored. The whole chromosome 4 painting probe hybridized on the entire chromosome of the abnormal chromosome 4 including the additional material (Figure 3A). Using telomeric probes, the presence of 4p and 4q telomeric regions was found on derivative chromosome 4 (Figure 3B). The Wolf Hirschhorn region (4p16.3) and centromeric probe revealed the presence of two signals on derivative chromosome 4 (Figure 3C). We concluded that the additional material of the aberrant chromosome was an intrachromosomal insertion duplication of 4q32-35; 46,XY,add(4)(p16.3).ish der(4)dup(4)(q32q35) ins(4)(p16.3q32q35)(wcp+,D4S3359+,D4S2930+,D4S96+,D4Z1+).
Discussion
Trisomy 4q syndrome was a rare condition, first described in 1971 [8]. The clinical findings are frequently presented including mental retardation, developmental delays, and multiple abnormalities such as microcephaly, acrocephaly, as well as malformed ears, a high/broad/depressed nasal bridge, and teeth and thumb anomalies. Genotype-phenotype correlation was difficult because of partial chromosome monosomy associated with 4q trisomy.
One boy with a duplication of 4q12-q13 was reported to have relatively few features, merely including microcephaly and mental retardation [9]. Likewise, the additional duplicated material from 4q25-q28 seems to produce no major clinical effects. The segment 4q27-4q31 seemed to be preferentially engaged in the trisomy 4q syndrome with severe clinical effects including growth retardation, mental retardation, microcephaly, facial asymmetry, thumb anomalies, hearing impairment, epilepsy, and a congenital heart defect [10]. Two familial cases of a small region duplication of 4q31.1-q32.3 and 4q31.3-q33 showed minimal clinical features [11,12]. Additional duplicated 4q34 material seems to be without major clinical effects, but telomeric band 4q35 may be involved in the development of microcephaly and severe mental and growth retardation [13]. Our patient with pure trisomy 4q32-35 was phenotypically normal except for azoospermia.
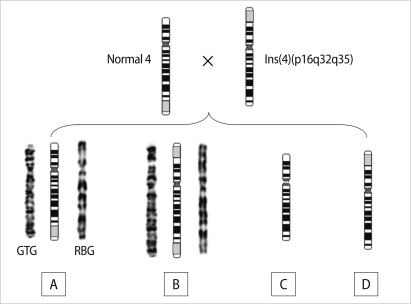
We wanted to confirm whether the meiotic segregation error appears in carriers with intrachromosomal insertion of chromosome 4 or de novo. Unfortunately, the chromosomal analysis of the parents was not available. However, we predicted that one of the parents might have a ins(4)(p16.3q32q35) because the der(4)dup(4q)ins(4) (p16.3q32q35) was a type of possible recombinant from pairing and crossing over during meiosis in carriers with the pericentric intrachromosomal insertion of chromosome 4 (Figure 4). Also, the risk of having a child with an unbalanced chromosome aberration from a carrier depends on both the type and size of the insertion. Individuals with a pericentric insertion are at a greater risk of having unbalanced offspring, but those with a paracentric insertion are at a relatively low risk.
To investigate rapidly whether the karyotype is balanced or unbalanced, CGH and FISH analyses can be a useful diagnostic tool. These techniques have been particularly useful in achieving an even higher resolution, such that small alterations (<3 Mb) in DNA copy number or location can be detected. Recently, array CGH analysis has been applied to the clinical diagnosis of chromosomal abnormalities and genomic aberrations [14].
In conclusion, we reported a case with an unbalanced karyotype of pure trisomy 4q32-35 which showed a normal phenotype except for spermatogenesis, though duplication often leads to some phenotypic abnormalities. Identification of the insertion is difficult. In this case, molecular cytogenetic analysis helped us in the precise detection. The precise characterization of an abnormal chromosome is clinically important for genetic counseling.