Introduction
Controlled ovarian hyperstimulation (COH) is routinely performed in assisted reproductive technology treatment, and frequently results in the production of surplus embryos. With the current trend to transfer only a single best-quality embryo to reduce the risk of multiple pregnancies, embryo cryopreservation is becoming a crucial component of assisted reproductive technology treatment. In patients at risk of developing ovarian hyperstimulation syndrome (OHSS), a safe option is to freeze all embryos to decrease the chance of severe pregnancy-associated OHSS. Currently, there is a trend to freeze all embryos and proceed with embryo transfer in nonstimulated cycles, as high levels of hormones from COH are believed to limit the implantation and pregnancy rate in fresh cycles by impairng endometrial receptivity [1,2].
Cryopreservation methods have developed and improved over the years. Currently, there are two major techniques: slow programmable freezing and vitrification. Vitrification is employed increasingly often, as it has many advantages over slow freezing [3,4,5]. For example, there is no ice formation and less chance of chilling injury [6], the embryos are outside the incubator for a shorter time, there is no need for expensive instruments, a minimal set-up time is required, and the post-warming survival rate is comparable to or even higher than that in the slow freezing method [7,8,9,10,11,12,13]. All stages of embryos can be vitrified, up to blastocysts [14,15,16,17,18,19,20]. It is predicted that slow freezing will be replaced entirely in the near future by vitrification for both human and industrial animal embryos [3].
During slow cooling, an embryologist can easily load a large volume of freezing medium containing the embryos into a straw, with no time constraints. In contrast, vitrification requires an embryologist to create a very small drop of vitrification medium (≤1 µL) that contains the embryos in a container within a time frame of ≤30 seconds. This process is very demanding and requires extensive practical skills. As a result, many embryologists are reluctant to switch from slow cooling to vitrification. The success of vitrification is also operator-dependent, and varies from laboratory to laboratory.
In this study, we developed an in-house freezing technique that combines the benefits of the vitrification and slow cooling methods. Embryologists familiar with the slow freezing method can immediately switch to our method without the need to acquire new skills. The objective of this study was to compare our method with the widely employed Cryotop vitrification method, using two-cell mouse embryos as a model. The outcomes were the immediate survival rate post-cryopreservation, further cleavage and blastocyst formation rates, and cell number in the blastocysts.
Methods
Outbred Institute of Cancer Research (ICR) mice were purchased from the National Animal Institute, Mahidol University, Bangkok, Thailand. They were kept at the Animal Husbandry Unit, Faculty of Medicine, Chiang Mai University, in a well-ventilated room at 25℃±2℃, under 60%–70% humidity, and controlled 12-hour light/dark cycles. We closely followed international and national guidelines for ethical conduct in the Care and Use of Animals for Research [21]. The Institutional Animal Care and Use Committee of the Faculty of Medicine, Chiang Mai University approved the use of mice in our study (protocol no. 39/2526). Before the experiment, we kept the mice undisturbed for 5 days to avoid the effect of stress from transportation.
1. Collection of two-cell embryos
ICR female mice, 5–7 weeks old, were superovulated by an intraperitoneal injection of 10 units of pregnant mare serum gonadotropin (Sigma, St. Louis, MO, USA), followed 48 hours later by an intraperitoneal injection of 10 units of human chorionic gonadotropin (Pregnyl; MSD, Bangkok, Thailand). Immediately after the second injection, superovulated females were paired with 9- to 20-week-old ICR males. They were checked for mating by the presence of vaginal plugs 16 hours later. Approximately 36 hours after the second injection, the mated female mice were sacrificed by dislocation of the cervical vertebrae. Two-cell embryos were flushed from the oviducts with phosphate-buffered saline solution (PBS; Gibco, New York, NY, USA), containing 0.5% bovine serum albumin (BSA, Sigma). Embryos were washed twice in equilibrated cleavage medium (COOK, Brisbane, Australia) under paraffin oil (Medicult, Jyllinge, Denmark). Only two-cell embryos with an intact zona pellucida and equal blastomeres with no fragmentation were selected for the experiments.
2. Interventions
Two-cell embryos were randomly allocated into three groups: a non-frozen control group (group 1), a group that underwent Cryotop vitrification (group 2), and a group that underwent our in-house method of freezing (group 3). In group 2, the embryos were vitrified and warmed using a Cryotop commercial kit (Kitazato BioPharma, Tokyo, Japan), following the manufacturer's recommendations. In group 3, the embryos were transferred into 100 µL of cryopreservation medium, containing 10% ethylene glycol, 5% propylene glycol, and 3% BSA in PBS with 0.2 M trehalose at room temperature (25℃). After 5 minutes, the embryos were loaded into 0.25 mL straws using the same method as in slow programmable freezing, with the end sealed with polyvinyl alcohol powder. The straws were inserted into the holes of a custom-made aluminum cylinder, which was precooled in liquid nitrogen for 20 minutes before use (Figure 1). The aluminum block only acted as a passive cooling device, and was very reliable if it was submerged at least 70% in liquid nitrogen for >10 minutes. After 5 minutes, the straws were removed, loaded into a cane, and stored in liquid nitrogen for 1 week.
There were four thawing solutions (TS1, TS2, TS3, and TS4), containing 1 M, 0.5 M, 0.25 M, and 0 M trehalose, respectively, with 0.3% BSA in PBS. They were warmed to 37℃ before use. Thawing was done by immersing the straws into a warm water bath at 37℃, until the content of the straws became clear. The straws were wiped with alcohol and both ends were cut with a pair of sterile scissors. The content of the straws were emptied into a Petri dish. The embryos were located, and transferred with minimal volume into TS1, TS2, TS3, and TS4 for 5 minutes each, then washed and cultured.
3. Embryo culture and assessment of embryo development
Surviving embryos were cultured in groups of 10 in 10-µL drops of cleavage medium under paraffin oil in an atmosphere of 6% CO2, 5% O2, and 89% N2 at 37℃. After 48 hours, embryos were washed in blastocyst medium (COOK), and transferred into the corresponding drops of blastocyst medium under paraffin oil.
Embryo development was assessed under an inverted microscope (Nikon, Tokyo, Japan) after 24, 48, and 72 hours of culture. Mouse blastocysts were classified as early, partial, full, expanding, hatching, and hatched blastocysts, using the criteria proposed by Gardner et al. [22] for human blastocyst development. In this study, only expanding, hatching, and hatched blastocysts were considered to represent good-quality blastocysts.
4. Differential staining of inner cell mass and trophectoderm cells
Differential staining was performed on all hatching and hatched blastocysts, using the protocol described by Pampfer et al. [23]. In brief, the blastocysts were washed three times in calcium- and magnesium-free buffer, before exposure to rabbit anti-mouse antibody (Sigma M5774; concentration 1:50) for 30 minutes at 37℃. After washing, they were transferred into guinea pig complement serum (Sigma S1639) with propidium iodide (Sigma P4170) and bisbenzimide (Sigma B2261) at 37℃ for 10–15 minutes. The blastocysts were washed and transferred onto glass slides to allow air drying. The slides were mounted in glycerol, and the number of cells in the inner cell mass (ICM) and the trophectoderm (TE) were counted using a Nikon E600 epifluorescence microscope, equipped with the LUCIA FISH program (Laboratory Imaging, Prague, Czech Republic). The ICM nuclei were observed to stain blue, while the TE nuclei showed an intense pink color. Mitotic nuclei were counted as one piece, but dead or pyknotic nuclei were not counted.
5. Statistical analysis
We used STATA ver. 8.2 (StataCorp, College Station, TX, USA) for statistical analyses. The chi-square test was used to compare the survival rate, further cleavage rate, and blastocyst formation rate in the three groups. The mean numbers of the ICM and the TE cells of the blastocysts were compared by one-way analysis of variance (ANOVA), with the Scheffé post-hoc test as appropriate. Two-tailed p-values <0.05 were considered to indicate statistical significance.
Results
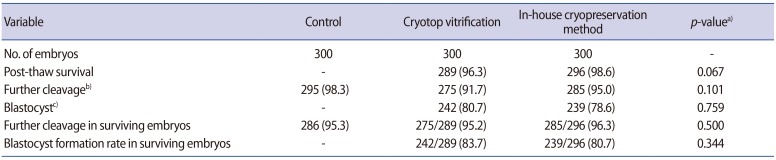
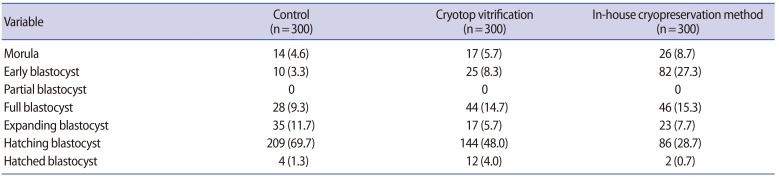
Nine hundred two-cell mouse embryos, with equal blastomeres and without fragmentation, were included in the study. There were no significant differences between the two cryopreservation groups in the immediate survival rate after cryopreservation, the further cleavage rate, or the subsequent blastocyst formation rate (Table 1). However, the further cleavage rate and blastocyst formation rate were significantly lower in both cryopreservation groups than in the non-frozen control group (p<0.05) (Table 1). The numbers of embryos in various stages of development after 72 hours of culture are shown in Table 2. The number of good-quality blastocysts was significantly higher in the control group (248/300, 82.7%; p<0.001) than in the Cryotop vitrification group (174/300, 58.0%), which in turn was higher than in our inhouse freezing group (111/300, 37.0%; p<0.001).
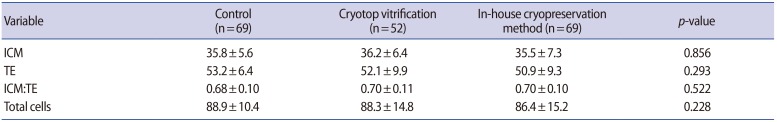
There was no significant difference in the mean number of cells in the ICM (ANOVA test; p=0.856), TE (ANOVA test; p=0.293), or in both the ICM and TE (ANOVA test; p=0.228) in blastocysts in the control group and the two cryopreservation groups (Table 3). Likewise, no significant difference was found in the ICM to TE ratio (ANOVA test, p=0.522) in blastocysts in the three groups.
Discussion
Mouse models are often employed to optimize techniques for human embryo cryopreservation, as the results are usually applicable to human embryos with little or no modification [24,25]. These models also avoid the ethical and legal issues of experimentation on human embryos. In this study, the two-cell stage was selected because it is a suboptimal developmental stage that is considered to be more sensitive to cryodamage than other stages of cleavage [26,27].
Vitrification has now emerged as the cryopreservation method of choice for oocytes and embryos of all developmental stages [7,28]. However, many embryologists still prefer slow cooling to vitrification because of their past experiences with the technique. Vitrification requires a long learning curve to master the skill of transferring an embryo in a microdrop (≤1 µL) of vitrification medium into a cryocontainer in 30 seconds or less. Recently, an automated vitrification device (Gavi, Genea Biomedx, East Kent, UK), which requires little if any intervention or sampling handling, was introduced to overcome human factors and to improve the efficiency of vitrification [29]. However, the device is expensive, and requires special consumables to operate, making the system unaffordable for many in vitro fertilization centers in developing countries.
In this study, we developed an in-house freezing method that combined the advantages of slow cooling and vitrification. The embryos could be loaded into a straw in a relatively large volume as in slow cooling, and then inserted into a hole inside a precooled aluminum cylinder as in vitrification. The whole procedure could be completed in approximately 10 minutes, and did not require expensive equipment.
In our hands, Cryotop vitrification yielded a survival rate and a further cleavage rate comparable to other studies (96.3% vs. 90%–100% and 91.6% vs. 93%, respectively) [30]. For our in-house freezing method, the survival rate (98.6%), further cleavage rate (95%), and blastocyst formation rates (78.6%) were comparable to those achieved using Cryotop vitrification (96.3%, 91.6%, and 80.7%, respectively). However, at 72 hours after cryopreservation, the embryos that underwent Cryotop vitrification developed into hatching blastocysts at a higher rate than those that underwent our in-house freezing method, but still at a significantly lower rate than those in the control group (47.9%, 28.7%, and 69.7%, respectively; p<0.001). However, it was reassuring that the mean numbers of cells in the ICM and TE, as well as the ICM-to-TE ratio, in the hatching and hatched blastocysts in the two cryopreservation groups and controls were comparable.
In Cryotop vitrification, embryos were vitrified within 30 seconds after exposure to vitrification medium. This method is known as nonequilibrium vitrification because the concentration of cryoprotective agents (CPAs) inside and outside the cells does not reach equilibrium. In contrast, in our freezing method, we employed a lower concentrations of CPAs (15%), and allowed a longer exposure time, as this concentration of CPAs was less toxic to cells than the vitrification medium. After equilibration, the straw containing the embryos was cooled in an aluminum cylinder previously immersed in liquid nitrogen.
In our system, we found that the temperature inside the straw decreased from 25℃ to −130℃ in about 10 seconds (a cooling rate of −930℃/min). It was likely that microscopic ice crystals formed in the extracellular compartment. Conversion of extracellular water into ice increased the concentration of the solute in the unfrozen solution around the cells, which drew water out of the cells along the osmotic gradient. As a result, the solute concentration in and around the cells became so high that water in the solution could not freeze. Due to the fast cooling rate, the glass transition temperature (Tg; around −130℃) was reached before the microscopic ice crystals had time to recrystallize into fewer but larger ice crystals that were lethal to the cells. The cells survived freezing because they were confined to unfrozen spaces between growing ice crystals [31].
The fact that the warming and cooling rates decrease when the final temperatures are approached creates another problem for rewarming cryopreserved samples because the final warming temperature is 37℃, while the final cooling temperature is −196℃. Therefore, by Fourier's law, the rate of warming through the critical zone of ice nucleation and formation will be slower than the rate of cooling through this zone [32]. Previously, it was presumed that CPAs exerted their main effects by preventing ice crystallization during cooling. It is now realized that cells can tolerate a certain degree of intracellular and extracellular ice reasonably well, and that devitrification can occur even during conventional vitrification [32]. Vitrification, in itself, is not a prerequisite for cell survival. Instead, the choice of CPAs is very important for vitrification, as they vary in their ability to limit ice recrystallization during warming and in the degree to which they enhance cells' tolerance of intracellular ice crystals [32]. In our study, we tried many different combinations of CPAs before ending up with the final mixture by trial and error.
Although our new freezing method was still inferior to Cryotop vitrification, we believe that it could be improved and would have advantages over the current vitrification methods, as it required a lower concentration of CPAs, making it less toxic to cells, and the time and volume of embryo loading were less critical than in the prevailing systems [33,34,35,36]. There are many possible ways to improve our freezing system, such as changing the base medium from PBS to a 1:1 mixture of PBS and HEPES, as this mixture is known to stabilize pH better than PBS alone at low temperatures [37]. Antioxidants could also be added to the freezing and thawing media, as cryopreservation can generate reactive oxygen species that are harmful to cells [38]. The level of serum albumin or trehalose in the media could be increased to increase viscosity and to facilitate water removal from cells. Finally, antifreeze substances in our system might be advantageous by preventing the formation of large ice crystals during warming [32].
A drawback of our study was that we used blastocyst quality as the final outcome. A better method would be to transfer post-cryopreservation embryos into pseudopregnant mice, and then count the number of implantation sites in each uterine horn. Moreover, data obtained from our mouse model might not be directly applicable to humans. In conclusion, our new freezing method is promising and deserves further study. It has the potential to be applied for clinical use in human embryo cryopreservation.