Introduction
Hydatidiform mole (HM) is sporadic and androgenetic diploid in origin, and represents the most common type of gestational trophoblastic disease. These pathologic conceptions are thought to result from fertilization of an oocyte where maternal chromosomes have been lost and endoreduplication of a single sperm genome occurs (or fertilization by two sperm), yielding a diploid DNA content derived entirely from the paternal genome [1]. Rarely, HM can be recurrent or familial [2,3]. Detailed homozygosity mapping and gene mutation screening in these settings has identified two causative loci, 19q13.4 and 6q13—chromosomal regions which contain NLRP7 and KHDC3L, respectively [4]. Of note, approximately 70% of women affected by familial recurrent HM have autosomal recessive mutations of NLRP7 [5]. Compelling evidence that an abnormal oocyte is responsible for the pathophysiology of recurrent HM is provided from observations that in vitro fertilization (IVF) cycles using donated oocytes for women with NLRP7 mutations result in normal offspring [6]. Nevertheless, several questions remain open regarding this phenomenon: (1) is the NLRP7 protein and/or its corresponding ribonucleic acid (RNA) required in the oocyte, and, if yes, at what stages; (2) what are the consequences of recessive NLRP7 mutations for the oocyte and for the embryo (cultured in vitro) which derives from it; and (3) when do embryo developmental defects manifest in this setting? This case report contributes new information addressing each of these unresolved issues.
Methods
1. Clinical presentation and genetic assessment
A 35-year-old G5P0050 presented with her husband for reproductive endocrinology consultation. Both partners were of Latino heritage and in excellent health. Her blood type/Rh group was A positive; she had no known drug allergies; and the patient took no medication on a regular basis. Between 2001 and 2012, the couple established five unassisted conceptions, all of which were HM and electively terminated in the first trimester. After the initial pregnancy, additional evaluation discovered that the patient had an European Society of Human Reproduction and Embryology Class IIb uterine anomaly/bicornuate uterus although her medical records did not define which uterine compartment contained the pregnancy for all cases. To evacuate all molar tissue, each event was followed by dilation and curettage performed by a gynecologist in an outpatient setting. However for her most recent pregnancy in 2012, serum human chorionic gonadotropin (hCG) levels did not decline normally after surgery and the patient received nine cycles of intravenous. doxorubicin [7] for low risk gestational trophoblastic disease under co-management with an oncology team. She was then placed on oral contraceptive pills, referred to a genetic counselor and advised not to conceive again for 12 months.
2. Molecular characterization of parents at NLRP7
The couple expressed interest in IVF and preimplantation genetic screening (PGS) of embryos before transfer. Anonymous donor oocyte IVF was also discussed but this option was declined as first-line treatment for personal reasons. While basic chromosomal data (standard G-band karyotypes) from both partners had been reassuring, further evaluation and counseling was obtained before moving forward with IVF in April 2016. Written informed consent for clinical and research aspects of the proposed treatment were obtained from both partners, and the investigational component was approved by our laboratory's independent institutional review board. The rare nature of recurrent HM [3,8] was discussed and genomic sequencing for the NLRP7 gene was carried out for both individuals. Briefly, genomic DNA was extracted from blood samples collected by peripheral venipuncture and, after evaluating the quantity and quality of the DNA, nucleic acids were disrupted to mean fragment size of 250 bp. Hybrid capture was used to enrich for areas of interest that included the coding and splicing regions of the NLRP7 gene. Pair-ended DNA sequences were obtained by next generation sequencing (NGS) using the NextSeq platform (Illumina, San Diego, CA, USA) at a mean depth of 100X; all regions of interest were characterized by coverage equal to or greater than 20X. Analysis of deletions and duplications was derived from NGS data; potential deletions or duplications were excluded for the index case.
3. Follicular recruitment, IVF protocol, and fertilization technique
Pre-IVF screening tests were normal for both partners. Ovarian reserve [9] was considered satisfactory by serum anti-Müllerian hormone and antral follicle count estimates (4.63 ng/mL and n=12, respectively). Monophasic oral contraceptive pills were used for suppression in the cycle immediately prior to IVF. No ovarian cysts >15 mm diameter were detected and baseline serum estradiol was <80 pg/mL. The patient received a combined follicle-stimulating hormone +human menopausal gonadotropin regime and gonadotropin-releasing hormone (GnRH)-antagonist commenced on stimulation day 6 [10,11]. Estradiol and follicular monitoring via transvaginal ultrasound continued until at least three follicles had maximum diameter of ≥17 mm. This threshold was achieved after a 12 day follicular recruitment interval, and 2500 IU hCG+80 u GnRH-agonist (leuprolide) was administered subcutaneous. with oocyte retrieval scheduled 36 hours later. Because PGS was planned, fertilization for all oocytes was exclusively via intracytoplasmic sperm injection (ICSI) [12].
4. Embryo culture, biopsy, and modified PGS protocol
Embryos were initially cultured in 10 µL microdrops of G1:3 culture medium (Vitrolife, Goteborg, Sweden), and on d3 post-fertilization placed in 20 µ of G2:3 (Vitrolife) to support blastocyst culture. As all embryos in this study had six or more cells by d3, each was placed in 20 µL of G-MOPS medium (Vitrolife) under mineral oil and subjected to biopsy following zona ablation via noncontact laser (ZILOS-tk; Hamilton Thorne Inc., Beverley, MA, USA). Cells from each embryo were removed using micromanipulation and placed immediately in RNAse–DNAse-free polmerase chain reaction (PCR) tubes containing 10 µL of PCR-grade water. Following addition of 50 µg/mL Proteinase K (Roche Biochemicals, Mannheim, Germany) each sample was placed under oil and transferred to our laboratory. DNA assessment used the Illumina Infinium HumanKaryomap-12 DNA Analysis kit. Beadchips were read by the iScan array scanner. Data were analyzed with BlueFuse Multi software to determine familial haploblocks. The screening protocol involved a first-round multiplex PCR, simultaneously amplifying the coding region of NLRP7 along with two unlinked fluoroscopically labelled (Cy5.5) microsatellite markers, GABRB3, and D13S314. The first round PCR reaction was then subject to (1) analysis of NLRP7 genotypes using real-time PCR protocol and allele-specific hybridization probes using the LightCycler system 1.0 (Roche Diagnostics, Manheim, Germany) and (2) microsatellite sizing on a Visible Genetics OpenGene System automatic DNA sequencer with Gene Objects software (Visible Genetics, High Wycombe, UK) to exclude chance contamination, as described previously [13].
Results
1. Mutation analysis
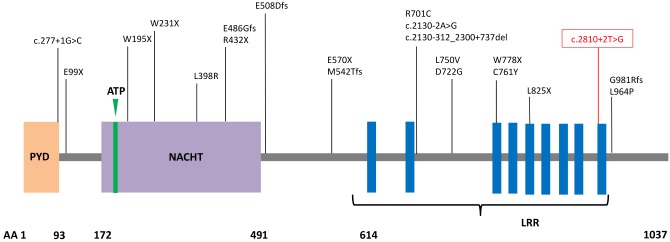
Variants in the NLRP7 gene were annotated following American College of Medical Genetics guidelines for interpretation. A substitution affecting the invariant donor splice site at position c.2810+2T>G of the coding region was identified in the sample obtained from the wife (Figure 1), while no pathogenic or likely pathogenic variants were identified in the husband. This pathogenic variant was shown to alter splicing of intron 9 and result in abnormal retention of the intron, which in turn inserts 54 amino acids in the protein (Figure 2). Of note, this pathogenic variant was previously reported in three patients with recurrent HM, all of Latino-American origin [14]. Our patient was fully informed and counseled about all results when available, and elected to proceed with IVF, PGS, and potential single embryo transfer.
2. IVF response, embryology data, and PGS findings
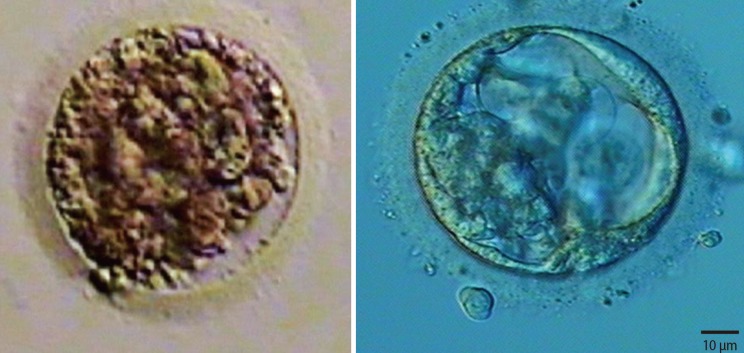
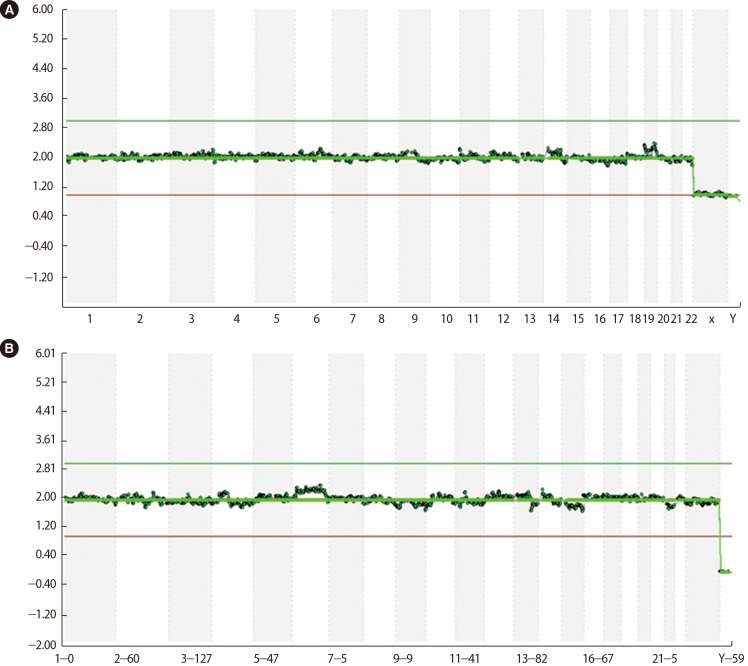
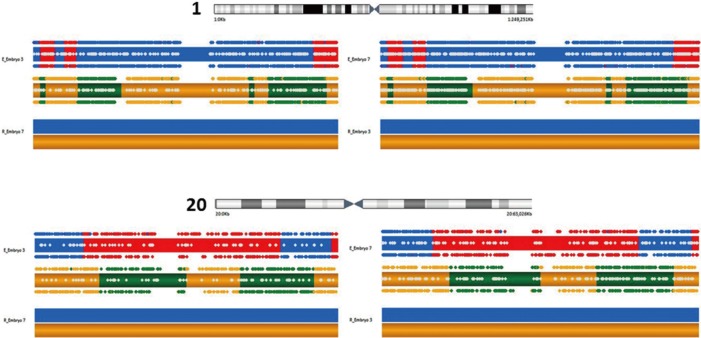
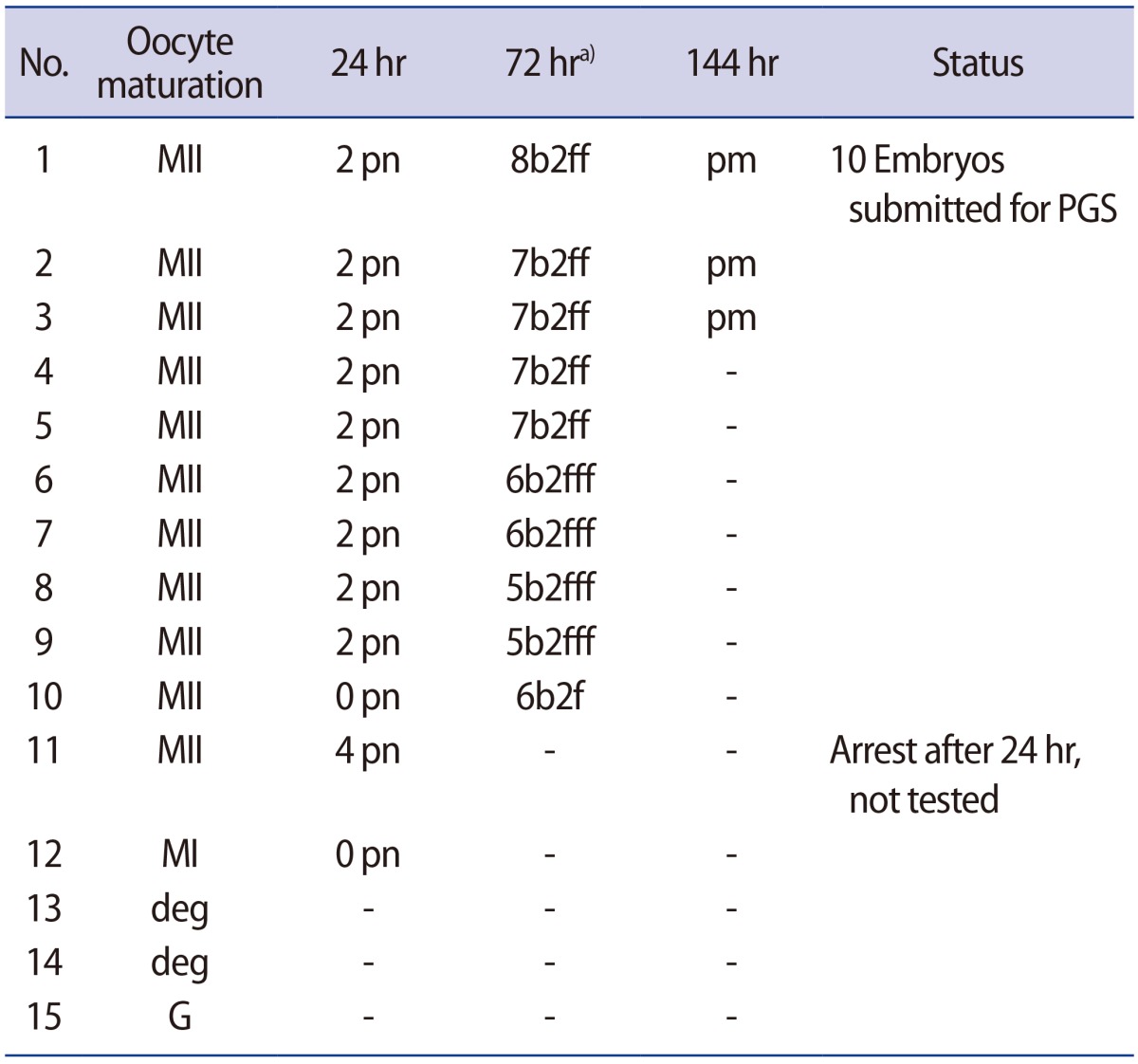
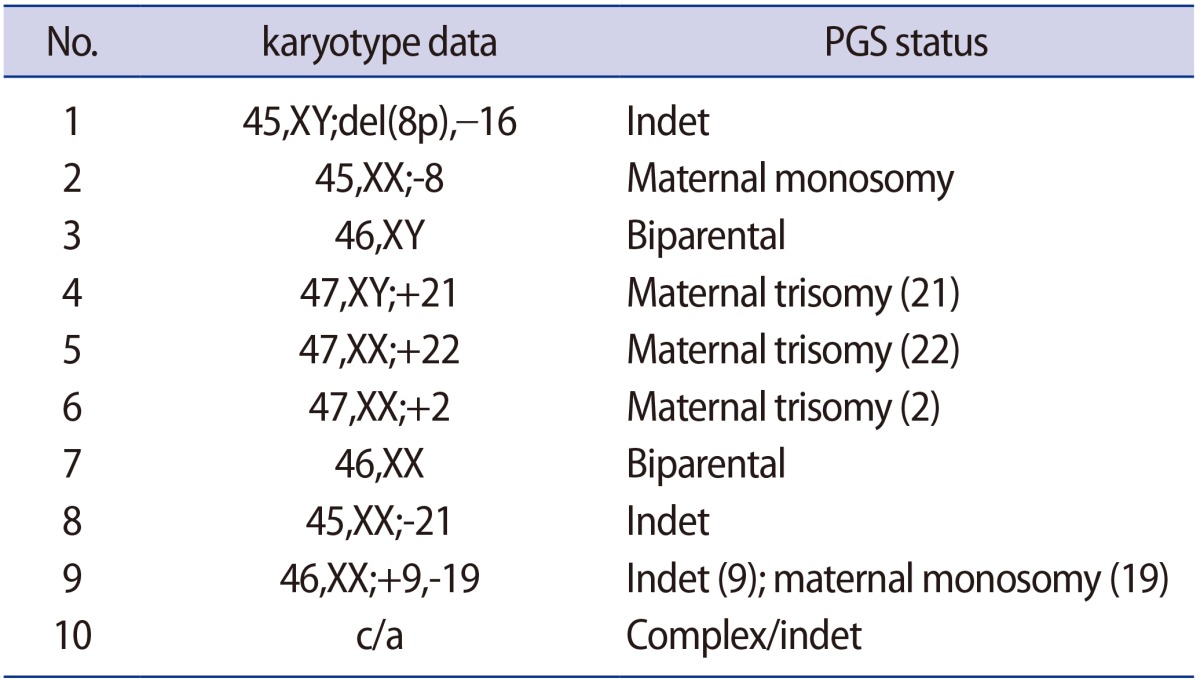
Fifteen oocytes were retrieved and nine progressed to 2 pronuclei (pn) stage following ICSI. An additional oocyte (initially designated as 0 pn) cleaved after 72 hours and joined the others as a provisionally viable zygote. However, arrest of all embryos was noted between 72 and 144 hours in culture (Figure 3), rendering none suitable for transfer. Progression of embryo development and associated morphology is summarized in Table 1. The 10 non-viable embryos were submitted for molecular diagnosis and biparental chromosomal contributions were identified in all embryos. While ploidy error for some embryos was of indeterminate origin, none was of paternal origin while five were of maternal origin (Table 2). A summary of NGS findings and genetic recombination events observed in the two euploid embryos is presented in Figures 4 and 5.
Discussion
NLRP7 is a maternal-effect gene believed to be required in oocytes to sustain normal development of the early human embryo. Previous research has shown that assisted reproductive technologies incorporating donor oocytes can correct the rare problem of recurrent HM [6]. Within oocytes, NLRP7 localizes to the cytoskeleton and is abundant in the cortical region [16]. However, the actual consequence of a mutation at NLRP7 as monitored during in vitro embryo culture has not been directly observed and reported until now.
For our patient, her history of five consecutive HM pregnancies implicated a genetic etiology for her condition. As with the previously reported mutation at c.2810, our patient with recurrent HM in California was also of Latino ethnicity, although our case was homozygous for this mutation while the earlier analysis identified a compound heterozygous state [14]. While our report is the first to describe development of 2 pn zygotes and abnormal fertilization after ICSI (as well as continued albeit variable development among observed embryos), we can only speculate why some embryos arrested sooner than others. The role of our patient's uterine anomaly in the context of recurrent HM is probably coincidental; to date, there has only been one published case describing the coexistence of bicornuate uterus and HM [17]. As our patient was known to harbor a homozygous mutation at NLRP7, all embryos generated were expected to inherit one abnormal copy of the gene responsible for HM. With a large enough denominator of screened embryos available for screening, we hypothesized that at least one euploid embryo or blastocyst might enable a transfer and subsequent opportunity for healthy pregnancy given this autosomal recessive disorder. Two euploid embryos were indeed identified for this couple after comprehensive chromosomal screening, but unfortunately both embryos arrested in culture and no in utero transfer was possible. This observation raises questions about the reasons behind this developmental collapse.
NLRP7 subcellular localization has been explored using an ex vivo-stimulated peripheral blood mononuclear cell model, revealing that NLRP7 co-localizes with the Golgi apparatus and the microtubule organizing center. Interestingly, a defect in microtubule and actin filament organization was suggested by Edwards et al. [18] to explain postzygotic abnormalities during in vitro culture of embryos from a patient with recurrent HM. A maternal-effect model has thus been proposed to explain the transcript abundance of NLRP7 which accumulates in developing oocytes and in early preimplantation embryos [19]. Such expression profiles suggest an involvement in the control of maternally-derived epigenetic programing or early developmental events in the zygote that occur ahead of embryonic genome activation. It has also been proposed that a mutation at NLRP7 may interfere with expression of imprinted genes on other chromosomes [20]. Dysfunction of such processes could explain why global embryonic arrest was observed in our case. Thus it may be that the presence of only one copy of this mutation at NLRP7 is sufficient to interrupt normal human embryo development.
It may be inferred that maternal NLRP7 mutations impair cytokine secretion by affecting their trafficking either through the classical Golgi-mediated secretory pathway or by disrupting the microtubule network known to play a role in the non-classical pathway of interleukin-1β secretion. In contrast, paternal transmission of NLRP7 mutations does not interfere with spermatogenesis, as males homozygous for NLRP7 mutations can father children [21,22]. How such a maternal mutation might manifest in the context of human embryo development has not been directly observed until now. The earlier hypothesis predicting developmental arrest secondary to a NLRP7 mutation is validated by the current case, which graphically highlights the catastrophic disruption of human embryo growth when fundamental cytoskeleton elements are disabled due to this specific mutation.
In contrast to these findings from preimplantation embryos, genotyped molar tissues from pregnant patients with recessive NLRP7 mutations are diploid biparental and do not have aneuploidies. The observed genetic differences between IVF embryos and chorionic villi of HM might be explained by the fact that HM tissues are genotyped with multiplex microsatellite markers, a technique less comprehensive than karyomapping and unable to detect all aneuploidies. Perhaps more crucially, molar tissue is typically sampled during the first trimester and may have been subject to preferential selection or growth of cells without ploidy error, a phenomenon well documented in fetal loss where aneuploidy rates decline with advancing gestational age.
As oocyte donation is being considered by our patient now, her prognosis for a healthy pregnancy with IVF and single embryo transfer is bright. In the future, patients with this NLRP7 mutation should be advised about the potential for cycle cancellation due to embryo arrest, and donor egg/embryo IVF or adoption encouraged as preferred management.