A case of the rare variant of Klinefelter syndrome 47,XY,i(X)(q10)
Article information
Abstract
Klinefelter syndrome is the most common genetic form of male hypogonadism, but the phenotype becomes evident only after puberty. It is characterized by infertility, small testes, sparse body and facial hair, increased body weight, gynecomastia, increased LH and FSH, and a low level of testosterone. Early recognition and treatment of Klinefelter syndrome can significantly improve the patient's quality of life and prevent serious consequences. Here, we report an infertile man with a rare variant of Klinefelter syndrome with a 47, XY, i(X)(q10) karyotype.
Introduction
Klinefelter syndrome is the most common genetic form of male hypogonadism, but the phenotype becomes evident only after puberty [1]. It is characterized by infertility, small testes, sparse body and facial hair, increased body weight, gynecomastia, increased LH and FSH, and a low level of testosterone [2]. Genetic counseling for these patients is important considering the progressive nature of hypogonadal features in Klinefelter syndrome. When serum testosterone concentrations in patients with Klinefelter syndrome become low, life-long substitution therapy is indicated and should be started as early as possible to avoid symptoms and sequelae of androgen deficiency. Early recognition and treatment of Klinefelter syndrome can significantly improve the patient's quality of life and prevent serious consequences.
About 80% of cases of Klinefelter syndrome are due to a supernumerary X chromosome (47,XXY) and the remaining 20% have 46XY/XXY mosacism or higher grade sex chromosomal aneuploidies (48, XXXY, 48XXYY, 49XXXXY) [3-5]. Here, we report an infertile man with a rare variant of Klinefelter syndrome with a 47, XY, i(X)(q10) karyotype.
Case report
The 33-year-old patient, who had been born to a 25-year-old woman and her 30-year-old husband, was referred to a male infertility clinic due to azoospermia. The patient generally looked healthy with a normal male appearance. His height was 160 cm, body weight 53 kg, and body mass index 20.7 kg/m2. There was a normal male pattern of body hair, and he had no gynecomastia. No mental retardation was observed. Examination of the external genitalia revealed both small testes to be normally positioned, but on palpation they had a soft consistency and a volume of 5 mL each. There were no abnormalities of the epididymis or the vas deferens and no signs of varicocele.
The family history of infertility was negative, and the fertility tests for his younger brother were normal. From two semen analyses by centrifugation, he was diagnosed with complete azoospermia. Endocrinologic investigation revealed elevated FSH and LH levels with FSH 69.04 mIU/mL (normal range, 1.27-19.26 mIU/mL), LH 22.17 mIU/mL (normal range, 1.24-8.62 mIU/mL), prolactin 26.48 mIU/mL (normal range, 2.64-13.13 mIU/mL), and testosterone 2.31 mIU/mL (normal range, 2.80-8.00 mIU/mL).
Metaphase spreads from the lymphocytes were prepared and trypsin-Giemsa banded according to standard techniques. His karyotype revealed 47,X,i(Xq),Y on 50 metaphase chromosomes with no mosaicism, and all of the isochromosomes were apparently monocentric (Figure 1). The microdissection for testicular sperm extraction was negative, and the histology of a testes biopsy revealed severe tubular atrophy showing only a few atrophic seminiferous tubules showing Sertoli cells only with Leydig cell hyperplasia (Figure 2). Genetic counseling about the disease was performed, and the possible usage of donor sperm was discussed for the fertility problem. Androgen replacement therapy was also recommended to avoid symptoms and sequelae of androgen deficiency.

Metaphase karyotype analysis showing isochromosome Xq.
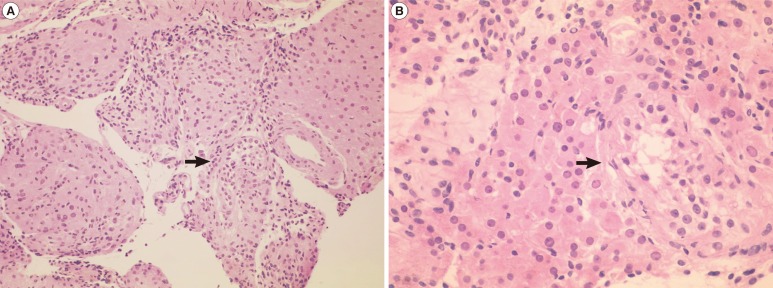
A testis biopsy shows severely atrophied seminiferous tubules. (A) Only a few atrophic seminiferous tubules (arrow) are present with Leydig cell hyperplasia (H&E, ×100). (B) An atrophic seminiferous tubule (arrow) showing Sertoli cells only (H&E, ×200).
Discussion
Klinefelter syndrome, first described in 1942 as an endocrine disorder, is the most common sex chromosomal aneuploidy disease and occurs in one of every 500-600 newborn males. The typical karyotype of Klinefelter syndrome is 47,XXY, but chromosome mosaics with 46,XY/47,XXY and complements with multiple X chromosomes like 48,XXXY are known [3-5]. The 47,X,i(Xq),Y in our case is a very rare variant form of Klinefelter syndrome. Isochromosome Xq is a structural rearrangement frequently observed in Turner syndrome, but it is apparently rare in males [6]. It is suggested that the most probable origin of an Xq isochromosome is misdivision of the centromere or sister chromatid exchange of one X chromosome. Due to the limited number of cases, the prevalence of this Klinefelter syndrome is still unclear. Up to the present, 21 patients with i(Xq) have been reported in the literature, and reports on Klinefelter syndrome with an isochromosome Xq have been discussed briefly in the literature; therefore, genetic counseling is difficult for such cases [7].
In general, all 47,X,i(Xq),Y patients have been reported to exhibit the main clinical features of Klinefelter syndrome, including reduced androgenization, small testes, azoospermia, gynecomastia, and elevated FSH and LH levels. In Klinefelter syndrome, when testosterone serum concentrations in patients are low, lifelong substitution therapy is indicated and should be started as early as possible to avoid symptoms and sequelae of androgen deficiency. For fertility issues, a testicular sperm extraction procedure may be considered for any viable sperm before androgen replacement therapy. It is well-documented that 47, XXY males show a mean height of about 6.5 cm more than their male relatives [8]. The only clinical difference between the 47, XXY and 47,X,i(Xq), Y Klinefelter syndrome patients is normal-to-short stature in the latter. The height of the patient in our case is compatible with the previous reports. The delineation of the phenotype of subjects with isochromosome Xq Klinefelter syndrome is important to provide meaningful genetic counseling. However, due to the rarity of this disease, only further evaluation with a larger group of such patients would be able to determine more clearly the clinical features and appropriate genetic counseling for these patients.
Notes
No potential conflict of interest relevant to this article was reported.