Epigenetics: A key paradigm in reproductive health
Article information
Abstract
It is well established that there is a heritable element of susceptibility to chronic human ailments, yet there is compelling evidence that some components of such heritability are transmitted through non-genetic factors. Due to the complexity of reproductive processes, identifying the inheritance patterns of these factors is not easy. But little doubt exists that besides the genomic backbone, a range of epigenetic cues affect our genetic programme. The inter-generational transmission of epigenetic marks is believed to operate via four principal means that dramatically differ in their information content: DNA methylation, histone modifications, microRNAs and nucleosome positioning. These epigenetic signatures influence the cellular machinery through positive and negative feedback mechanisms either alone or interactively. Understanding how these mechanisms work to activate or deactivate parts of our genetic programme not only on a day-to-day basis but also over generations is an important area of reproductive health research.
Introduction
Epigenetic aberrations have been conjectured to be highly relevant to sexual and reproductive health, as they account for the interactive relationship among the genomic landscape, gene environment interactions and disease phenotype. Novel insights into aetiologies of complex non-Mendelian disease traits have provoked a burgeoning interest in the field of reproductive epigenetics. How a range of epigenetic mechanisms can differentially influence the male and female germ line and developmental process is being closely scrutinized. Of late, the subtle and elegant modulation of fidelity of transmissible heritable characteristics through epigenetic reprogramming has also received wider scientific attention. Developmental activation and deactivation of epigenetic signatures at the pre-implantation phase provides putative links between assisted reproductive technologies and imprinting disorders. Occurrence of imprinting errors disrupts placental growth and development in assisted conception procedures.
It is important to understand how epigenetic information is established and regulated in the parental germline and how epigenetic inheritance takes place. This crucial knowledge may be of help to determine the connection between environmentally manipulated epigenetic alterations and development of the organism. It has often been argued that most epigenetic modification, by whatever mechanism, is erased with each new generation, during gametogenesis and after fertilization. DNA methylation, histone modification, microRNA (miRNA) expression, and nucleosome positioning are the four basic modes driving the path of development (Figure 1). Epigenetic control systems generally involve three types of proteins: "writers," "readers," and "erasers (Table 1)." Writers attach chemical marks, such as methyl groups (to DNA) or acetyl groups (to the histone proteins that DNA wraps around) [1]. Readers bind to these marks, thereby influencing gene expression; erasers remove the marks. The marks are passed down as cells divide, providing a sort of cellular memory to ensure that cell proliferation is effectively regulated.
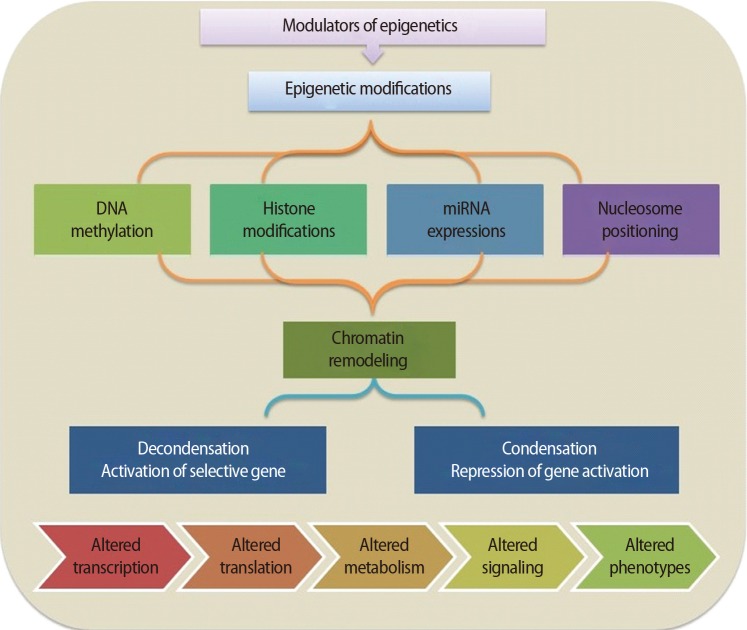
The basic mechanisms of epigenetic regulation. miRNA, microRNA.
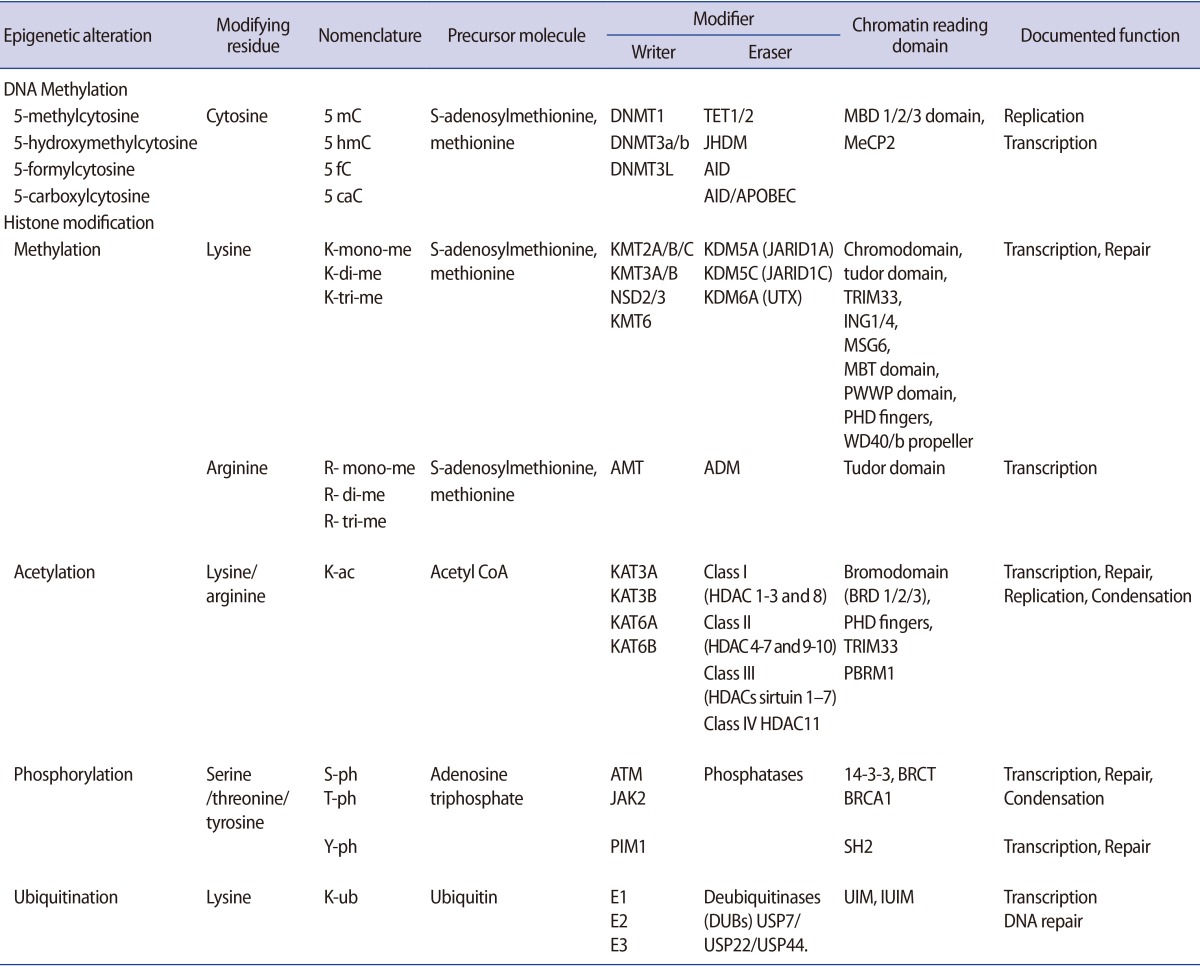
Molecules involved in the process of epigenetic regulation
1. Epigenetic modifications
1) DNA methylation
DNA methylation, a well-characterised epigenetic modification, is a heritable covalent modification and is binary in nature. Most methylation occurs at the number five carbon of the cytosine pyrimidine ring (5-methyl-cytosine or 5 mC) and represents less than 5% of all cytosines in our genomes. Genomic methylation patterns are propagated during cell division by DNA methyl-transferases (DNMT1, DNMT3A/B), which catalyse the transfer of a methyl group to DNA [2]. Five DNMT enzymes—DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L—actively regulate two different processes, that is, maintenance and de novo methylation activities [3]. One of the important sites for DNA methylation is CpG-enriched regions associated with promoters called "CpG islands [45]." The majority of these sites are located in the promoter region and first exon of genes. DNMTs, along with other enzymes, can orchestrate gene silencing and maintain a repressive chromatin state. Once methylated, proteins such as MeCP2, MBD1, MBD2, and MBD4, which have a methyl-binding domain (MBD), bind to the DNA, which further impedes recruitment of transcription factors to DNA, leading to abrogated gene expression [6]. Hypermethylation of DNA can also recruit histone deacetylase (HDAC), leading to inevitable alterations in gene expression [78]. DNA demethylation, a related event that is equally important, converts methyl-cytosine into cytosine either actively or passively. The replication-independent active mechanism uses the ten-eleven translocation (TET) enzyme family (TET1, TET2, and TET3) to catalyse hydroxylation of 5mC followed by activation-induced cytidine deamination [910]. On the other hand, replication-dependent passive demethylation occurs during inadequate availability of global methyl donor S-adenosyl-L-methionine (SAM) or DNMTs-mediated demethylation in the presence of Ca2+ ions and reducing surroundings [11]. Thus, promoter hypermethylation causes gene silencing whereas promoter demethylation results in gene expression.
2) Histone modifications
Histones are the globular proteins that undergo posttranslational modification and alter regulation of gene expression, and DNA replication, recombination, and repair. While nucleosomes represent the primary step in the construction of higher-order chromatin structures [12], histones have a protruding charged 15-38 amino acid N-terminus ("histone tail") that influences nucleosome assembly into higher order chromatin structures. In its condensed state, chromatin remains in a folded configuration so that the nucleosomes are stacked, and hence, not readily accessible to gene activation. However, covalent modifications such as acetylation, methylation, phosphorylation, poly-ADP ribosylation, and ubiquitination on the long tails of histones alter histones-DNA interaction and higher order chromatin folding. These post-translational covalent modifications regulate the contact between the octamer core and DNA, and determine DNA accessibility to transcription factor complexes. The capability to accumulate information appears to dwell in the amino-terminal tails of the four core histones, which are exposed on the nucleosome surface and are subject to enzyme-catalysed post-translational modifications of select amino acids, including lysine acetylation, lysine and arginine methylation, serine or threonine phosphorylation, lysine ubiquitination, lysine sumoylation, or glutamine ADP ribosylation (Table 1). Epigenetic modification of the histone tail plays a key role in transcriptional regulation, DNA repair, DNA replication, alternative splicing, and chromosome condensation. With reference to its transcriptional state, the human genome can be approximately compartmentalised into actively transcribed euchromatin and transcriptionally inactive heterochromatin [13]. Euchromatin is characterised by high levels of acetylation and trimethylated H3K4, H3K36, and H3K79. On the other hand, heterochromatin is categorized by low levels of acetylation and elevated levels of H3K9, H3K27, and H4K20 methylation [14]. Interestingly, such dynamic modifications are actively added or removed by different histone-modifying enzymes (writers and erasers, respectively) that catalyse modification of specific amino acids with definite modifying groups in a site-specific manner to manage transcriptional control (Table 1). With specific modification, histones regulate the structural organization of chromatin by altering the electrostatic charge provided by the substituted group and facilitating recognition sites for different adaptor proteins (readers) such as proteins containing a bromodomain, which binds to the acetylated lysine inscriptions.
3) miRNA
MicroRNAs (miRNA) are single-stranded RNAs approximately 21–23 nucleotides in length that are transcribed from DNA but not translated into proteins. These non-coding RNAs (ncRNAs) regulate several molecular pathways at the transcriptional or post-transcriptional level. miRNA genes primarily reside between genes (intergenic) or within introns (intronic) of genes and are transcribed to a primary miRNA (pri-miRNA) mediated by polymerase II or III [15]. The pri-miRNA is processed within the nuclear compartment to a precursor miRNA (pre-miRNA) by Drosha, a class 2 RNase III enzyme. Subsequently, the transport of pre-miRNAs to the cytoplasm is arbitrated by exportin-5. In the cytoplasmic region, they are processed further to develop into mature miRNAs by Dicer, an RNase III type protein, into 21–25 nucleotide double-stranded RNAs (dsRNAs) and loaded onto the Argonaute (Argo) protein to generate the effector RNA-induced silencing complex (RISC) [16]. RISC binds to mRNA, forming a RISC-mRNA complex on which miRNAs mediate sequence-specific recognition and binding, which further degrades or silences the target mRNA depending on the sequence complementarity. While the majority of researchers believe that miRNAs restrain translation, evidence that miRNAs can actually augment translation through alterations in the Argo component of the RISC has also been reported. Thus, while miRNAs appear to police translation in an inhibitory fashion, they may also boost translation in defined biological settings.
4) Nucleosome positioning
DNA packaging in nucleosomes might affect all stages of transcription, thereby regulating gene expression. The precise position of nucleosomes around the transcription start sites has an essential degree of control over the initiation of transcription [17]. Nucleosome positioning not only decides the accessibility of the transcription factors to their target DNA sequence but has also been reported to take part in shaping the methylation landscape. Besides transcription regulation, nucleosome occupancy also participates in directing meiotic recombination events. The precise function of nucleosomes is influenced by the incorporation of different histone variants that are incorporated into chromatin independently, outside the S-phase. Often linked with specific histone modifications, nucleosome remodelling machinery is also influenced by DNA methylation. Thus, the interaction among diverse epigenetic partners is often evident.
2. Role in gametogenesis
Gametogenesis occurs in a precisely defined microenvironment, and therefore molecular events during this process have to be strictly regulated to enable correct transmission of heritable information to subsequent generations. Primordial germ cells (PGCs) are the derivative source of both female and male germ cells that experience genome-wide reprogramming during their relocation to the developing gonads. Reprogramming in the germ line is a crucial event to retune parent-specific epigenetic information, and is potentially vital for organization of sex-specific germ line development and identity [18]. The processes of development from germ cells to gametes and from gametes to embryos include dramatic cellular differentiation accompanied by drastic alteration in gene expression that occurs through tight regulation by genetic as well as epigenetic mechanisms [18]. These epigenetic mechanisms may be involved in checking meiosis and the terminal differentiation programme during gametogenesis, retaining information in gametes for the offspring and erasing improper marks in zygotes before the beginning of a new being. The chromatin structure of germ cells acts as a basic mechanism to preserve their unique self-renewal ability and block differentiation. In addition to chromatin remodelling, dynamic regulation of histone modifications and DNA methylation is also required to maintain germ cells' identity [19]. Developmental epigenetic characteristics are established in the process of gametogenesis and early embryogenesis (Figure 2).
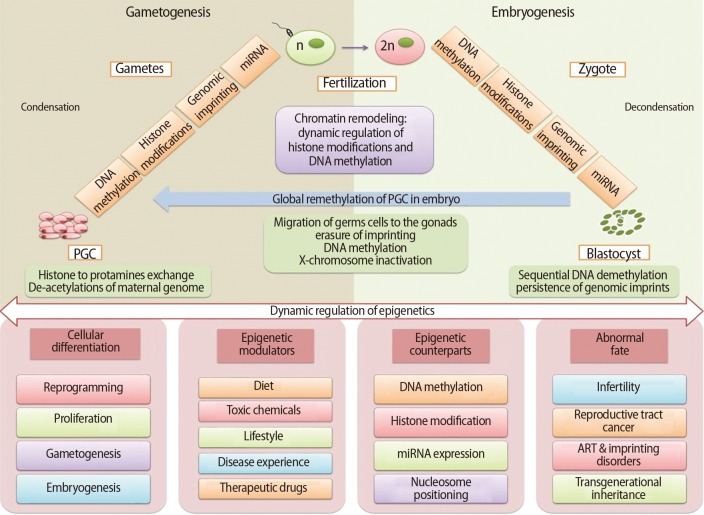
A summary of epigenetic mechanisms during gametogenesis and embryogenesis. PGC, primordial germ cell; ART, assisted reproductive technology.
1) Spermatogenesis
Male germ cells have compact nuclear DNA which are required for the transmission of the paternal genome to the oocyte. The highly compacted paternal DNA residing in the sperm head goes through extensive remodelling [20]. The process of spermatogenesis involves condensation of chromatin in the spermatid head prior to conversion of spermatids to spermatozoa [21]. The spermatids have a less compact genome in the early stages of spermatogenesis that is further compacted in the sperm genome by the substitution of histones by non-histone proteins. In the process of histone replacement, they are first replaced by transition proteins (TNP1 and TNP2) and eventually by protamines (P1 and P2). This allows the protamines-bound sperm genome structure to compact 6 to 20 times more than the histone-bound nuclear structure that makes it transcriptionally and translationally inert. Human spermatozoa are acknowledged to enclose a huge range of RNA molecules, including over 100 types of miRNA [22]. On the other hand, the mature sperm cells enclose only the mRNA and small RNAs that were present in the spermatids at the early phases of spermatogenesis. It was previously assumed that there is no participation of sperm transcriptome in embryo development but several recent studies have described the involvement of sperm genome organization as well as paternal transcriptome in early embryonic development. Moreover, chromatin of mature spermatozoa retains small amounts of histones, which is crucial in the differentiation and early development of the zygote. Surprisingly, the male pronucleus also displays elevated levels of histone acetylation, which supports higher transcription from the S phase in the zygote stage and later. Demethylation in the PGCs results in activation of genes: deleted in azoospermia-like (Dazl) and synaptonemal complex protein 3 (Sycp3), that are essential for gametogenesis [23]. This also regulates expression of testis-expressed (Tex19.1, Tex19.2) and Mili (also called Piwi-like 2) genes required to suppress any unmasked retrotransposon action and for a stable genome integrity [24]. miRNAs also regulate the transcriptional silencing in compact chromatin-containing elongating spermatids [25]. Dicer activity has been found to be crucial during male germ cell differentiation, as reported in Dicer knockout studies, which shows that early Dicer deletion results in damage accumulation and compromised spermatogenesis [2627]. Increasing evidence supports the vital role played by small RNA-mediated RNA regulation in normal spermatogenesis and can affect male fertility if it fluctuates from the normal germline.
2) Oogenesis
The maternal DNA contained in the oocyte is bound by histones acquired during oocyte growth comprising post-translational modifications associated with stalled metaphase II. The major difference between the chromatin of oocyte and somatic nuclei is the deficiency of H1 linker histones in oocytes, which are replaced with a specific H1 variant whose role in early embryogenesis remains to be understood. Histone methylations play important roles in the regulation of chromatin structure and gene expression during follicle maturation, especially for oocyte meiotic maturation. Euchromatin histone-lysine N methyl-transferase 2 (EHMT2) methylate H3K9me1 and H3K9me2 are crucial for early meiotic progression. It has been established that H3K9me3 appears in growing oocytes from early antral follicles, increases subsequently during the growth phase, and is retained during oocyte meiotic maturation and activation [2829]. However, at different developmental stages, sensitivity toward methylation of a diversity of histones including H3K4me, H3K4me2, and H3K4me3 has been found in granulosa cells of follicles as well as in oocytes from primary to antral-stage follicles [30]. In addition, histone ubiquitination may be another regulatory factor during follicle development. Ubiquitinated histone H2A is coupled with transcriptional silencing of large chromatin areas during meiosis in females. The production of gametes might require orderly and proper epigenetic reprogramming in premigratory and migratory germ cells for an appropriate epigenotype to support subsequent normal development. Genomic imprinting in the oocytes of females occurs after birth and is arrested at the diplotene stage of meiotic prophase I, and is then completed by the de novo methylation process in the fully-grown oocyte stage.
3. Role in embryogenesis
Organisms experience two developmental epigenetic reprogramming events: the first during gametogenesis and the second in the pre-implantation embryo [31]. Embryonic development includes an array of cell fate decisions to specify cell lineages that are established early during development, and that must be "maintained" through multiple cell divisions. It is increasingly evident that epigenetic inscriptions play a critical role in this cell memory during development [32]. It has long been appreciated that the parental pronuclei in mammalian zygotes and during preimplantation development acquire asymmetric epigenetic information, which include distribution of histone modifications and differences in the level of DNA methylation [3334]. At this stage, the two parental genomes stay physically separate and undergo a diverse program of chromatin remodelling. Epigenetic restructuring is essential for transition of a totipotent zygote into pluripotent stem cells, leading to channelization to the multipotent lineage progenitor cells that finally reach the lineage-specific unipotent somatic cells. The stem cell transcriptome reprogramming by epigenetics is the ultimate control mechanism that provides stem cells with adaptability, flexibility, and versatility so they can modify their gene expression in response to developmental signals and differentiate into any cell (Figure 2). Here, the epigenetic modifiers play an important role to drive the information into cell-specific functional lineages [35]. Reversible DNA methylation and covalent histone modifications are two important agents in these mechanisms, where transient histone modifications provide short-term flexibility in the early development of lineage-committed genes. Besides this, the various stages of development can be forced into long-term or permanent repression by DNA methylation, nucleosome positioning, and higher-order chromatin reorganization [36]. Recent reports from numerous laboratories have provided evidence that the recently fertilized oocyte acquires epigenetic signals from the sperm chromatin that are necessary for proper embryonic development [37].
After the fertilization of highly specialized ova and sperm, a wave of demethylation takes place in the zygote genome that continues till the 16-cell stage of development. The processes of epigenetic remodelling occur differently in the genome of each parent at this stage. The paternal genome goes through numerous cycles of decondensation by demethylation as well as protamines replacement with histones and histone variants. Following fertilization, the maternal genome completes meiosis and undergoes notably fewer epigenetic alterations as compared to the paternal genome. At the same time, imprinted regions escape demethylation cycles and maintain the specific epigenetic states of their parent of origin.
Reversible DNA methylation is an important epigenetic manipulator involved in a number of processes that maintain genome stability, organize mono-allelic expression of parentally imprinted genes, silence retrotransposons and confirm transcriptional silencing of genes on the inactive X chromosome. The two parental pronuclei have epigenetic asymmetry at the global levels of DNA methylation, as exhibited initially by high levels of DNA methylation [3338]. These asymmetric and persistent epigenetic marks in the early stages of zygotic development and their effect on early embryonic development remain unclear. DNA methylation patterns are established in embryonic development by DNMT3A/B [39]. Precise DNA methylation patterns are required for regular development and lineage commitment. This causes the methylation of the entire genome while CpG islands are protected, which causes global repression and permits housekeeping gene expression in all cells. Although DNA methylation event is secondary, it possibly contributes an additional level of repression by providing long-term stability. However, it can be cut off both actively and passively at the episodes of reprogramming in PGCs and preimplantation embryos [40].
DNA demethylation is a very important player in epigenetic reprogramming of embryonic development and differentiation that determines cellular fate [4142]. At the time of development, germline DNA methylation marks are globally removed in the blastocyst and a bimodal pattern is reestablished later during implantation when the entire genome gets methylated while CpG islands are protected. Just after fertilization, DNA-demethylation triggers the quiescent transcriptional machinery of the totipotent zygote which helps throughout germline reprogramming of the PGCs and eventually propels the pluripotent stem cells into lineage-restricted pathways. Both active and passive mechanisms of DNA demethylation mediate remodelling of the paternal genomes just after fertilization and before the initial zygotic divisions. Genome-wide active demethylation in the paternal pronucleus occurs before pronuclear fusion and first cleavage division [43]. Active demethylation involves extensive oxidation followed by passive loss over early cell divisions in the zygote [44]. Towards the 16-celled morula stage it experiences a replication-dependent loss of methylation (LOM) of the maternal pronucleus [43]. The delineation of the trophoectoderm from the inner cell mass at the blastocyst stage is also facilitated by hypomethylation in the promoter region of transcription factor E-74-like factor 5 [45]. It is thought that DNA-demethylation excites the onset of pluripotency in the zygote, subsequently triggers the expression of lineage-determining transcription factors and determines the embryo's segregation into the three embryonic germ layers.
The next major reprogramming event in the developing embryo happens in germ cell precursors known as PGCs. These cells originate directly from the blastocyst's pluripotent cells of the inner cell mass. They migrate to urogenital ridges during gastrulation, where they are reprogrammed by erasure of all pre-existing epigenetic modifications such as removal of maintained marks that were made during the preimplantation stage [4647]. These mechanisms of regulation result in repression of somatic differentiation of genes and/or activation of genes concerned in preservation of specific germ cell identity [45]. These important phenomena therefore set up the sex-specific epigenetic profiles and transcriptional planning needed for the typical development of the germline and epigenetic inheritance regulation [48].
Reversible post-translational covalent histone modifications are crucial agents directing epigenetic regulation responsible for generation of differential transcriptional outcomes. Three different states of chromatin arrangement are seen depending on the occurrence and abundance of histone marks [49]. The first dynamic set of permissive marks, including tri-methylated histone H3 lysine 4 (H3K4me3), acetylated histone H3 lysine 9 (H3K9ac) and acetylated histone H4 (H4ac) are found in the totipotent zygote and pluripotent stem cells. In somatic cells, lineage control genes are stably silenced by repressive histone modifications such as trimethylated histone H3 lysine 9 (H3K9me3) and trimethylated histone H3 lysine 27 (H3K27me3), which are a second set. The third state of chromatin arrangement is a more important and exclusive feature known as bivalent domains, found in key developmentally regulated genes. This domain contains active H3K4me3 and repressive H3K27me3 along with repressive H2AK119Ub1 marks and unmethylated CpG DNA regions (non-mCpG) [50]. These chromatin mechanisms acts as molecular switches maintained at a poised state amenable for specific transcriptional response upon receipt of differentiation cues [1].
Histone variants also have specific role in reprogramming of the epigenome. This occurs after fertilization and contributes to the differences between the two parental genomes. The male pronucleus has been observed with a H3.3 histone variant that possibly plays a role in directing or preventing other paternal-specific histone modifications. Modification of H3.3 has also been linked with developmental arrest due to mutations in lysine 27 of H3.3 and paternal pericentric heterochromatinization [51]. Although the exact functions of these H2A.X and H2A.Z variants remains unclear in early development, their deletion brings developmental arrest and failure of implantation.
Throughout the cellular stages and stages of development, a cell's nuclear architecture undergoes extensive dynamic changes such as in the level of chromatin compaction, accessibility by transcription factors and nucleosome positioning within specialized nuclear regions. This affects the degree of condensation of chromatin and nucleosomal organization that allows region-specific transcriptional profiles to be established during the course of cell commitment [1]. Probably the most complete understanding of involvement of miRNAs in paternal epigenetic inheritance come from the studies in Caenorhabditis elegans [52]. Studies involving identification of RNA sequencing of sperm RNA also suggest the possibility that these RNAs could contribute to epigenetic states in the early embryo [53].
Genomic imprinting can occur at hundreds of coding genes and regulatory ncRNAs. This is one of the important phenomena in epigenetics that regulate gene activity by preferential allele-specific gene expression. Simply imprinting mono-allelic DNA methylation marks can control the expression of imprinted genes, which are than maintained throughout development in a lineage- or tissue-specific manner [54]. This specific epigenetic regulation leads to expression of only one parental allele of a gene in such a way that some imprinted genes exhibit paternal expression whereas others exhibit maternal expression. These imprints are obtained in the process of gametogenesis when genome-wide epigenetic remodelling occurs, which must be maintained throughout preimplantation development where another wave of genome-wide epigenetic remodelling occurs [55]. The basic mechanism of this process is DNA methylation-based molecular arrangement, which regulates the establishment and maintenance of parental imprints all over early embryonic development and gametogenesis [56]. It is imperative to mention that methylated DNA regions are transcriptionally inactive, whereas unmethylated DNA regions are transcriptionally active. Methylated DNA regions are inhibited from expression by two processes, a methyl group attached to DNA can interfere in the binding to a particular transcription and methylated DNA regions can recruit MBD proteins mediating transcriptional repression. Genomic imprinting and epigenetic reprogramming in the context of DNA methylation is governed by two major waves of genome-wide demethylation and remethylation. First, in biparental genetic totipotent zygotic cells just after fertilization, and second in biparental methylation in the differentially methylated regions (DMRs) that are eliminated, imprinted methylation is reestablished in the germline for the next generation. That means imprinted DMRs remain unaffected at this first wave of genome-wide DNA demethylation and maintain parental imprints in the somatic tissues of the embryo throughout life. Animal studies suggest that those imprints that are established in growing oocytes during primordial to antral follicle transition will remain unaffected in some genes until just prior to ovulation; which could be vulnerable to ovarian stimulation. Deviations in proper epigenetic reprogramming such as disruption of imprinting in the germline can endorse heritable changes on transcription and diseases.
4. Role in infertility
Changes in the germline genome and epigenome can be a path for environmental and evolutionary adaptations. However, aberrant epigenetic remodelling of the germline is proposed as a potential mechanism by which gametogenesis can be compromised and can result in fertility and reproductive health-related problems. Emerging evidence suggests that genetic factors (cytogenetic abnormalities, DNA damage, disease status, hormonal abnormalities, effect of the micro environment, etc.) and environmental factors can have harmful effects on epigenetic arrangement during the process of implantation, placentation, and foetal growth. Therefore, even in a genetically normal individual, the environment can induce epimutations (a heritable change in gene expression that does not affect the definite base pair sequences of DNA) and can result in infertility and subfertility not only in the parent's germline but also potentially in the offspring.
1) Male infertility
Results of recent work have significantly improved our understanding of the sperm epigenome and its probable role in embryonic development. These new outcomes have facilitated a broad definition of a normal epigenetic state in the male gamete while also providing insight into the potential aetiologies of various idiopathic male infertility cases [57]. Emerging studies have shown notable associations of aberrant DNA methylation in spermatozoa with idiopathic male infertility as well as increased frequency of spontaneous abortions and imprinting disorders [58]. In addition, the epigenetic errors in the process of spermatogenesis in humans have been acknowledged as causes for reduced sperm competency and reduced fertility in males. The fundamental causes of infertility in males may be seminal oxidative stress, double- and single-stranded DNA damage of both the nuclear and mitochondrial genome of sperm, sudden telomere attrition and aberrant epigenetic alterations that can not only affect a single person but also their descendants [59]. The relationships between DNA methylation levels and male fertility in humans have been investigated in several studies. The methylation load and defects of the sperm DNA have been confirmed as a cause of infertility. Emerging evidence suggest that miRNAs play an important role in male fertility. Specifically, miRNA knockout of Dicer in the sertoli cells has been shown to result in testicular dysgenesis and infertility due to alterations in sertoli cell architecture [60]. A concerted effort to identify the roles of particular miRNAs during spermatogenesis should help determine whether miRNAs can serve as important predictive or diagnostic indicators and/or a system for treating male infertility. Epimutations could lead to male infertility, as suggested in the literature. Often hypermethylation has been found to be associated with poor semen parameters or male infertility. Evaluating the implications of lifestyle habits, food intake and environmental factors (e.g., drugs and toxins) on the sperm epigenome and the subsequent effect on fertility outcomes is needed. This should be of help to better understand epigenome regulation in the male germline, and may also suggest new strategies for this line of research.
2) Female infertility
In females, epigenetic changes in the oocytes can be affected by factors such as maternal nutrition and exposure to certain toxins that have been linked with neonatal developmental and gestational defects [61]. Perturbations in epigenetic mechanisms may further elucidate the intrinsic causes of infertility or state of fertility or ovarian tumorigenesis [62]. Epigenetics in women with infertility is more complex and less explored; the factors associated with epigenetic abnormalities are diverse. These factors include ovulation problems (more common due to polycystic ovary syndrome [PCOS]), endometriosis, inflammatory disorders, age-related factors, and nutritional status [63]. PCOS is a complex multi-factorial, chronic disease state that is one of the leading causes of anovulatory infertility and subfertility. PCOS is distinguished by a range of ovarian, hormonal, and metabolic disturbances and exhibits chronic menstrual irregularities, follicular cysts on ultrasonography, hyperinsulinemia, obesity, and hyperandrogenemia. In utero hyperandrogenism exposure may perturb the epigenetic reprogramming in foetal reproductive tissue and could be a cause of post-natal PCOS. Endometriosis has not been proven to have a genetic cause, but it is worth considering that abnormal expression of candidate genes could be provoked by different epigenetic modifications including DNA methylation, heterochromatization or aberrant regulation of miRNA expression [64].
It is well established that the decline in female reproductive responses is associated with advanced maternal age and postovulatory aging of oocytes [65], which leads to compromised quality of oocytes and a lower pregnancy rate. Understanding the underlying causes of these circumstances, which may include mitochondrial dysfunctions, aneuploidy, or epigenetic changes, has recently drawn increased attention. This is an important factor in reproduction and reproductive health in order to ensure that the correct epigenetic modifications occur during oogenesis and early embryo development [66].
5. Role in assisted reproductive technology
Assisted reproductive techniques intended to support infertile couples in having their own children have significant risks of passing on molecular errors to the next generations [67]. Although assisted reproductive technology (ART) has become a common practice for the treatment of human infertility, the increased vulnerability of ART-conceived children to perinatal health problems is poorly understood [68]. According to the Centers for Disease Control and Prevention, "ART includes all fertility treatments in which both eggs and sperms are handled." These procedures include artificial insemination, in vitro maturation, in vitro fertilization, super-ovulation, embryo culture and transfer, and intra-cytoplasmic sperm injection [69]. ART is growing in popularity and becoming an increasingly important field. Currently, 1% to 3% of annual births occur through ART in industrialized countries and the proportion is growing [70]. These technologies are considered procedurally safe except for an increased incidence of premature births. In vitro culture and maturation of oocytes, superovulation, and embryo culture can stimulate epigenetic modification that might be transmitted to the next generation and expected to influence its epigenome [71]. These procedures affect the DNA methylation pattern, parental imprinting status, and expression of imprinted genes. Two important techniques employed during ART, ovarian stimulation and in vitro culture, play a crucial role in epigenetic programming events that happen naturally through gametogenesis and early embryonic development [72]. ART-related manipulations of oocytes and embryos coincide with the timing of epigenetic rewriting and sex-specific imprint acquisition. This suggests that ART can interfere with epigenetic programming events from this moment and beyond. In fact, studies show that imprinting disorders such as Beckwith-Wiedemann syndrome (BWS), Angelman syndrome and Prader-Willi syndrome are more common in children conceived by ART [72]. Several case series in children born via ART suggest that there is a three-fold to six-fold higher prevalence of ART-conceived children born with BWS than in the general population.
BWS is an overgrowth disorder where aberrant genetic and epigenetic regulation at the KCNQ1OT1 and H19 imprinted domains is the cause. Methylation abnormalities such as maternal LOM at KCNQ1OT1 and maternal gain of methylation at H19 have been identified in ART-conceived BWS children. Another common disorder seen is Angelman syndrome, which is a neurological disorder with genetic and epigenetic disturbances at the maternal LOM small nuclear ribonucleoprotein polypeptide N-imprinted domain due to downregulation of maternal UBE3A expression that cause abnormalities [73]. The major problem identified in these ART-associated imprinting disorders is due to epimutations that are usually absent in the normal population and maternal alleles [72]. BWS also arises from imprinting defects at the SNRPN DMRs.
Observed differences in DNA methylation levels in ART and in vivo conception are not only due to the underlying infertility but also the effect of some aspect of ART procedures themselves [74]. It should be noted that ART is generally performed in individuals with infertility/subfertility problems. As result, in most cases, successful ART produces the birth of a child that has abnormal expression due to improper imprinting in the parental lineages. This prompts the question of whether infertility/subfertility implies epigenetic reprogramming or whether the manipulation of gametes/embryos by ART leads to epigenetic alterations or whether a combination of both is involved [73]. It is possible that infertility and ART alone or in combination disrupt the complex biological pathways leading to aberrant epigenetic alterations such as the methylation status of imprinted regions and may cause imprinting disorders. However, the relatively small number of cases of ART associated with imprinting disorders makes it challenging to carry out conclusive clinical trials. Therefore, coordinated clinical and basic studies in large registries are mandatory to determine the actual impact of ART on epigenetic alterations. When the safety of human ART is considered from an epigenetic perspective, we must take into account the functional implications of epigenetic reprogramming in very early development and adult disease [6875]. Moreover, artificial conception process not only transmits sperm nuclear DNA to the oocyte but also activation factor, centrosomes, and a host of messenger RNA and microRNAs that can influence post-fertilization activity [76]. Children born of ART need to be evaluated with long-term follow-up from childhood into adulthood to identify potential impacts of genomic imprinting in infertility and ART [77]. As nutritional and/or metabolic status influences cellular microenvironment, therefore, both these factors might also influence the epigenomic landscape of children born from assisted reproductive procedures [78].
6. Transgenerational epigenetic inheritance
Epigenetic information could be transmitted to the subsequent generation as genetic information is passed on, and is now discussed as transgenerational epigenetic inheritance. This event can have a broad spectrum of implications for the state of health, disease development, and more importantly, molecular adaptation and evolution. The epigenetic transmission of information can occur due to connecting links, the gametes that can pass adhered epigenetic marks from the previous generation to subsequent generations. Because the environment can induce changes in the germline epigenome that can be transmitted to subsequent progeny, therefore, might be a cause for disease aetiology observed at later stages of life [798081]. The most common mechanism mediating transgenerational epigenetic inheritance is DNA methylation, by which the major programming occurs during early mammalian development [82]. However, ncRNAs have also been identified as a possible mechanism for regulating transgenerational transmission of information [83]. In addition, piRNAs have been noted as possible mediators of the mechanism that silences mobile genetic elements transgenerationally [8384]. Evidence suggests that transgenerational epigenetic inheritance includes the transmission of epigenetic marks between generations in the absence of any environmental exposure [85], but their limits, that is, the degree to which this can occur, remain unclear [86]. Therefore, identifying the mechanisms of this mode of transmission and their impact on life is an area of epigenomics waiting to be explored.
Direct exposure generally has an effect on somatic tissues, but a transgenerational effect requires a transmission of epigenetic information with the germline. It has been demonstrated that exposure in the parental generation is remembered and transmitted to unexposed future generations through gametes as a "memory" of the environment. Environmental and metabolic conditions during gestation may shape epigenetic reprogramming and may further influence the lifetime health of the child and the disease aetiology in offspring [878889]. This illustrates that the intrauterine environment (nutrition, stress, and so on) has a vital effect on developmental programming, which, in turn, not only influences the germline (sperm or egg) in the foetus (F1) but also the germline of the foetus (F2) [90]. When these influences are transmitted and continue to appear across generations beyond F3, this reflects the transgenerational inheritance of epigenetics and cannot be described by direct environmental exposure anymore. Evidence from further investigation has shown that epigenetic effects could be inherited into the fourth generation. However, due to versatility of exposure and the transient nature of epigenetics, it becomes more difficult to understand the mechanisms in this complex process [91]. It has now been shown in several different laboratories and animal model systems that stress can promote the epigenetic transgenerational inheritance of disease. The account of transgenerational disease and pathology has distinct and greater frequency than direct exposure pathology, as shown in recent studies [9293]. In this context, the epigenetic transgenerational inheritance of disease has drawn great interest and should be taken seriously in future health management and therapies. Research has shown that the exposure to environmental toxicants in the early postnatal phase can promote infertility [94]. Available molecular evidence has shown that epigenetic information carrier proteins in gametes play important roles in the transmission of phenotypes from parents to offspring [95].
Conclusion
Exploring and understanding the role of the epigenetic landscape in reproductive health is important. Diverse sets of consecutive epigenetic modifications form the basis of reproductive fitness (Tables 2, 3, 4, 5, 6, 7). The key reprogramming events in reproduction ensure correct establishment and maintenance of epigenetic marks in germ cell development and early embryogenesis, while preventing the transmission of faulty information to offspring. As epigenomic landscape could be influenced by nutritional and/or metabolic factors, both these factors might also influence the cellular microenvironment during development and later stages in life. Moreover, faults generated by missing critical steps in differentiation have consequences for infertility and imprinting disorders. Furthermore, the widespread performance of ART to treat infertility demands more thorough research from the epigenetic perspective, including a comprehensive strategy and planning to address nutrition, environmental factors, and in vitro embryo production. However, the potential for far-reaching transgenerational consequences requires exploration in suitable models.

Molecular epigenetic mechanisms in regulation of spermatogenesis

Molecular epigenetic mechanisms in regulation of oogenesis

Molecular epigenetic mechanisms in regulation of embryogenesis

Molecular epigenetic mechanisms in regulation of male infertility

Molecular epigenetic mechanisms in regulation of female infertility

Molecular epigenetic mechanisms in regulation of assisted reproductive technologies
While alterations in the epigenomic landscape are required for regular growth and development, they can also be responsible for some diseases. The significance of epigenetics in maintaining normal development and biology is reflected by the observation that many health complaints build up when the wrong type of epigenetic marks are introduced or are added at the wrong time or in the wrong place. Disrupting any of the four systems that contribute to epigenetic alterations can cause abnormal activation or silencing of genes. Thus, an organism might be prone to epigenetic reprogramming errors during the resetting of the genome of gametes and zygotes, which differentiate to create many specific tissue types. On the other hand, the reversibility of epigenetic marks suggests the possibility that the activity of key genes and pathways can be regulated as a therapeutic approach. This suggests that the epigenome has truly arrived as a target for drug development, as pharmaceutical companies and biotech giants have programmes aimed squarely at proteins that operate in the epigenetic space. Recent technological advances are enabling research on a genome-wide scale, which is facilitating a more "systems biology"-based approach to understanding disease aetiology.
Acknowledgments
The authors are thankful to the Department of Biotechnology, Council of Scientific and Industrial Research, and Department of Science & Technology, Government of India, New Delhi for providing financial support to the laboratory of Prof. Pradyumna Kumar Mishra.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.