Insights into granulosa cell tumors using spontaneous or genetically engineered mouse models
Article information
Abstract
Granulosa cell tumors (GCTs) are rare sex cord-stromal tumors that have been studied for decades. However, their infrequency has delayed efforts to research their etiology. Recently, mutations in human GCTs have been discovered, which has led to further research aimed at determining the molecular mechanisms underlying the disease. Mouse models have been important tools for studying GCTs, and have provided means to develop and improve diagnostics and therapeutics. Thus far, several genetically modified mouse models, along with one spontaneous mouse model, have been reported. This review summarizes the phenotypes of these mouse models and their applicability in elucidating the mechanisms of granulosa cell tumor development.
Introduction
Granulosa cell tumors (GCTs) are sex cord-stromal tumors that comprise 5% of all ovarian tumors in women [12]. Although GCTs arise mainly from granulosa cells, they can develop in both the ovaries in women and the testes in men [34]. GCTs are usually detectable at an early stage; however, 43% of patients experience recurrence, and 80% of those patients die from the disease [56]. Due to the indolent nature of these tumors, along with their propensity for relapse and malignancy, patients with GCTs need long-term follow-up to monitor whether recurrence or metastasis has occurred [78]. Inhibins have been used as reliable markers to diagnose GCT recurrence and progression [91011].
GCTs are classified into juvenile granulosa cell tumors (JGCTs) and adult granulosa cell tumors (AGCTs) based on histology, nuclear morphology, the age of occurrence, and the potential for disease recurrence. AGCTs are the most common type of GCT, and occur in peri-and postmenopausal women [12]. JGCTs occur in girls from infancy through puberty and have the potential for malignancy [7]. AGCTs often show prominent nuclear and histological features, such as nuclear grooves (coffee-bean nuclei) and Call-Exner bodies (small fluid-filled spaces surrounded by granulosa cells). By contrast, granulosa cells in JGCTs are neoplastic, round, non-grooved, luteinized, and have hyperchromatic nuclei [13]. Moreover, histological analysis shows the presence of follicle-like spaces in JGCTs.
Advances in the identification of molecular mechanisms implicated in AGCTs have identified the C402G missense mutation of the FOXL2 gene as present in 95% of AGCT patients [1415]. This mutation has not been found in JGCTs, and, furthermore, the loss of FOXL2 expression has been observed in aggressive JGCTs [16]. The absence of FOXL2 can alter the fate of granulosa cells, pushing them into uncontrolled growth, because FOXL2 expression is important for establishing and maintaining granulosa cell identity. The FOXL2C134W mutation may induce AGCT formation by regulating targets in apoptotic [17] and steroidogenic [18] pathways. However, no clear pathophysiological mechanisms have been described. While FOXL2 mutations in other loci induce mislocalization, protein aggregation, and impaired transactivation [19], the C402G missense mutation of the FOXL2 gene does not lead to alterations in FOLX2 protein subcellular localization, protein aggregation, mobility, or transactivational activity on its target promoter in vitro compared to wild-type protein FOLX2 [20]. Recently, it was proposed that GSK3β regulation on serine 33 (S33) of mutant FOXL2 is the cause of oncogenicity in AGCT [21]. Two activating mutations (R201C and R201H) of the stimulatory α subunit of a trimetric G protein (Gαs) were discovered in JGCT patients [22], and in-frame duplications within the pleckstrin homology domain of AKT1 were discovered in >60% of JGCT patients [23].
Two cell lines derived from human GCTs have been investigated to understand the etiology and molecular mechanisms of AGCTs and JGCTs [2425]. Despite the important information that has been obtained using these cell lines, some discordances have been observed with data obtained from studies of human tumors [26], suggesting that the use of mouse models for studying GCTs may be necessary to more fully understand their origins. Here, we summarize the phenotypes of the currently available GCT mouse models and what they have revealed about the molecular mechanisms underlying GCT development.
1. SWR mice
SWR/Bm (SWR) mice were reported in 1985 as a model for studying pathways leading to the formation of spontaneous JGCTs [27]. Approximately 1% of inbred female SWR mice develop malignant JGCTs at approximately 8 weeks of age, starting at the time of the first ovarian follicle maturation at approximately 3 to 5 weeks of age [28]. Possible tumor susceptibility modifiers include the Gct loci, such as Gct1 on chromosome 4 and Gct4 and Gct6 on the X chromosome. Gct1 is essential for GCT development and is responsive to the androgenic precursor dehydroepiandrosterone, which has been shown to increase tumor frequency [29]. Although other foci such as Gct2, Gct3, Gct4, Gct5, Gct6, Gct7, Gct8, and Gct9 are also linked with Gct1 and may be associated with the formation of GCTs, the Gct1SW allele is an essential driver for the ovarian tumor phenotype [30]. Four genes within the Gct1 interval (Vps13d, Tnfrsf8, Tnfrsf1b, and Dhrs3) may be involved in tumor formation in SWR mice. The tumors in this model are endocrinologically active, secreting high levels of inhibin and estrogen [29]. The initial formation of spontaneous GCTs is dependent on endocrine hormones, such as androgenic steroids at puberty, implying that the time frame of the first wave of maturing follicles is critical in the development of JGCTs. GCTs from SWR mice have neo-plastic potential, as demonstrated by the incidence of metastases through consecutive transplantation. SWR mice have significantly decreased serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH), as seen in human GCT patients, while inhibin-α is robustly increased [31]. The serum levels of progesterone, dihydrotestosterone, and testosterone are also reduced in SWR mice, while tumor-bearing mice have a high capacity for aromatization. Therefore, this mouse model has similar histological and endocrine characteristics to human JGCT patients.
2. Inhibin-α null mice
The inhibins belong to members of the transforming growth factor-β (TGF-β) family and inhibit the synthesis and secretion of pituitary FSH [3233]. These peptide hormones are expressed in the adrenal gland, pituitary, brain, spleen, kidney, central nervous system, placenta, and the gonads [32]. Targeted deletion of the Inha gene causes the development of gonadal stromal tumors as early as 4 weeks of age in both males and females, with nearly 100% penetrance [34]. Female mice develop multifocal, hemorrhagic, bilateral tumors with tubular or cord-like structures. Comparison of the serum FSH levels in these Inha-null mice shows a two- to threefold increase compared to heterozygous or wild-type controls, a characteristic that is secondary to the lack of suppression by inhibin. This suggests that downstream molecules in the inhibin signaling pathway are important for GCT formation, and imbalances in gonadotropins might also play a role [35]. As shown in follitropin receptor knockout (FORKO) mice [36], perturbations in the gonadotropin signaling pathway and milieu of the ovary induce the development of GCTs. FORKO mice have a high serum level of activin secreted from the ovarian tumor, resulting in a cachexia-like wasting syndrome that is lethal to the mice at the onset of ovarian tumor development [3437].
High levels of activin caused by elimination of the Inha gene induce activation of the SMAD2/3 signaling pathway in granulosa cells, stimulating proliferation [34]. The importance of SMAD3 for tumor progression is supported by studies of Madh3-/- (SMAD3-null) and Inha double knockout mice, which show slower progression of GCTs; SMAD2 is not necessary for inducing tumor formation in inhibin-deficient mice [383940]. Although inhibin-α null mice develop GCTs, their relevance for human GCTs is not completely understood because the majority of human GCT patients have high serum levels of inhibins [4142]. Nonetheless, this mouse model has been useful for understanding the downstream molecular pathways of GCT formation.
3. Mice with the simian virus 40 T-antigen fusion gene
Simian virus 40 T-antigen is a proto-oncogene that can transform cells. Transgenic mice with overexpression of the simian virus 40 T-antigen driven by the murine inhibin-α subunit promoter (Inha/Tag) were originally developed as a source of granulosa tumor cell lines to investigate the characteristics of GCTs. The ovarian tumors from these mice are prominent at 5 to 6 months of age and have 100% penetrance [43]. Tumor cells from these mice are atypical, mitotic, and have the appearance of granulosa cells, though the tumor cells cannot be classified as AGCT or JGCT. Additionally, a transgenic mouse line with overexpression of simian virus 40 T-antigen driven by the anti-Müllerian hormone (AMH) promoter also develops ovarian tumors [44]. The ovarian tumors are bilateral, with 10% of mice developing tumors at 3 to 8 months of age. These mouse ovarian tumors contain serous cystic spaces, large hemorrhages, and necrosis, showing further metastases in the lungs or liver in later stages. Cell lines derived from these mice maintain granulosa cell characteristics, such as the expression of LH, production of estradiol, responsiveness to human chorionic gonadotropin (hCG), and the presence of granulosa cell markers, as well as AMH type II receptors [44].
4. Mice with overexpression of the LHβ subunit (bLHβ-COOH-terminal peptide)
These mice were generated using bovine LH β-subunit/hCG β-subunit COOH-terminal peptide (bLHβ-CTP) [45]. Tumorigenesis in these mice occurs at 4 to 8 months of age. These transgenic mice show unusually high levels of LH, as well as precocious puberty, a prolonged luteal phase, formation of cysts and, thus, GCTs, causing infertility in transgenic female mice dependent on their genetic predisposition [46]. High levels of estradiol, testosterone, and progesterone are present, and LH is especially high, suggesting that elevated LH might contribute to the formation of GCTs. According to a report describing the crystal structure of hCG [47], LH has growth factor-like properties. Excessive levels of LH in transgenic mice result in angiogenesis and growth aberrations, indicating that abnormal gonadotropin stimulation is tumorigenic. However, these mice have many unique non-gonadal phenotypes due to chronically elevated steroid levels. The importance of excessive LH in inducing tumor formation is also supported by mice deficient in both inhibins and LH [48]. These mice show a delay in tumor progression and increased survival. Moreover, bLHβ-CTP and Inha/Tag double transgenic mice show much faster gonadal tumorigenesis with elevated serum levels of LH [49]. Altogether, these mice models indicate that excessive LH levels can induce tumor formation.
5. Mice with mutant Wnt/β-catenin
Ovarian granulosa cells express components of the Wnt/β-catenin pathway, suggesting that Wnt/β-catenin (CTNNB1) plays a role in granulosa cells. More specifically, both human and equine GCTs show nuclear localization of β-catenin. The PI3K/AKT signaling pathway can also activate the WNT/CTNNB1 pathway through inactivation of GSK3β [50]. Both Wnt4flox/-;Amhr2tm3(cre)Bhr/+ and Wnt5aflox/-;Amh2cre/+ mice are subfertile and have small ovaries, follicle atresia, and a decreased ovulation rate, indicating that the WNT signaling pathway is important in ovarian follicle growth/survival and steroidogenesis [5152535455]. Catnbflox(ex3)/+;Amhr2Cre/+ mice, which express stable nuclear β-catenin driven by the Amhr2 promoter, develop GCTs, suggesting the importance of the misregulated Wnt/β-catenin pathway in GCT development [56]. Follicle-like structures develop at 6 weeks of age, and in 57% of mice, the formation of GCTs occurs at 7 months. A microarray analysis of pretumoral aged mice showed high expression levels of Wnt/β-catenin antagonists and neuronal markers. These were localized in pretumoral lesions, implying that the misregulated Wnt/β-catenin signaling pathway changes the fate of granulosa cells [57]. Mice with Pten loss of Catnbflox(ex3)/+;Amhr2Cre/+ (Ptenflox/flox;Ctnnb1flo x(ex3)/+;Amh2Cre/+) demonstrate quickly growing GCTs with the ability to spread into the abdominal cavity, suggesting that PI3K/AKT and WNT/CTNNB1 signaling have synergistic effects in the development of GCTs [58]. Moreover, KrasG12D;Ctnnb1flox(ex3)/+;CYP19Cre/+ mice die earlier due to GCTs than Ctnnb1flox(ex3)/+;CYP19Cre/+ mice, indicating that the KRAS pathway is involved in GCT formation [4]. Therefore, misregulated WNT signaling in granulosa cells can affect cell development and the formation of GCTs.
6. Double conditional knockout mice of SMAD1 and SMAD5 in granulosa cells
Female mice deficient in both Smad1 and Smad5 in granulosa cells using Amhr2-cre are subfertile and develop GCTs with 100% penetrance [59]. This mouse model shows a phenotype at 2 to 3 months that is similar to that of humans with JGCTs [60]. Furthermore, 80% of these mice develop peritoneal and lymphatic metastases by 8 months of age [59]. This mouse model suggests that a signaling pathway involving the activation of SMAD2/3 or the disruption of SMAD1/5 is conducive to JGCT pathogenesis. SMAD1/5 double-knockout mice had lower serum levels of FSH and altered LH and estradiol levels than control animals. Moreover, serum levels of inhibin-α and AMH are highly elevated in this mouse model. WNT/β-catenin in SMAD1/5 double-knockout mice is not significantly different from that observed in wild-type mice, suggesting that WNT/β-catenin may not contribute to JGCTs in the SMAD1/5 mouse model. Recent reports have demonstrated that TGFβ-SMAD signaling contributes to JGCT development through a study of Smad1/5/4 triple-knockout mice, which were found to exhibit delayed tumor formation and no evidence of metastasis, in contrast to Smad1/5 double-knockout mice [61]. These findings suggest that the TGFβ signaling pathway contributes to tumor formation in JGCTs through the repression of apoptosis.
7. Mice with double-mutant BMPR1A and BMPR1B in granulosa cells
The BMP signaling pathway is important for granulosa cell development. Among the type I receptors of the BMP signaling pathway, BMP receptors 1A and 1B are expressed in granulosa cells [62], and the knockout of both genes results in GCTs [63]. Tumors from Bmpr1a/1b double-knockout mice show upregulated TGFβ and TGFβ target genes. The ovaries of Bmpr1a/1b double-knockout mice develop bilateral ovarian tumors from 8 (≤40%), 16 (≤90%) months of age. The gene expression profiles of ovarian tumors of Bmpr1a/1b double-knockout mice are similar to those of Smad1/5 double-knockout mice tumors, although some differences between the two exist. This implies that the BMP signaling pathway through the BMP ligands, BMPR1A/1B to SMAD1/5, is important for the regulation of tumor suppressor pathways in mouse granulosa cells.
8. Estrogen receptor-β knockout mice
Pituitary and ovarian tumors are observed in estrogen receptor (ER)-β knockout mice female mice at 2 years of age. GCTs in ERβ-/- mice secrete estrogen and have high expression of ERα [64]. Pituitary tumors induce the high expression of gonadotropin-releasing hormone, which consequently causes the proliferation of granulosa cells, as well as endometrial hyperplasia, resulting in ovarian tumors [64]. Regarding the role of ERα in ovarian tumor formation, Couse and Korach [65] showed that 40% of ERα/β double-knockout female mice developed sex cord-stromal ovarian tumors between 15 and 20 months of age. However, Fan et al. [64] showed that ERα/β double-knockout female mice did not develop ovarian tumors, emphasizing the necessity of ERα in the development of GCTs. Consistent with this observation, female ERα-/- and Inha-/- double-knockout mice show an enhanced onset of GCT formation, shorter survival, and induced hypergonadotropism caused by disruption of the negative feedback mechanism in the absence of ERα [66]. Burns et al. [66] also showed that ERα was the genetic modifier involved in the development of ovarian tumors using ERα/Inha double-knockout mice and ERα/β/Inha triple-knockout mice. However, the survival curves for ERβ/Inhadouble-knockout mice overlapped with those of Inha mice, indicating that ERβ alone is not enough to induce tumor formation. High expression of the LH receptor and SMAD3 are seen in both ERβ/Inha double-knockout and inhibin-α knockout mice. Therefore, ER signaling pathways may have protective effects on tumor formation in females, with acceleration of tumor formation upon mutations in the ERα locus and the loss of both ERα and ERβ.
9. Mice with depletion of Foxo1/3 and Pten
The selective inactivation of the Foxo1 and Foxo3 genes in mouse ovarian granulosa cells leads to the development of GCTs in ≤20% of Foxo1/3 double knockout mice by 6 to 8 months of age. Although Pten conditional knockout mice with loss of the Pten gene in granulosa cells (Ptenf/f;Cyp19-Cre) show persistent nonsteroidogenic luteal cells [67], some mice (1%–7%) develop GCTs in Ptenf/f;Amhr2-Cre [58]. Additional inactivation of the Pten gene in the Foxo1/3 strain enhances the onset of GCT formation to 65% in the Foxo1/3/Pten triple-knockout mice at approximately 2 to 3 months of age [68], suggesting that the loss of Pten in the Foxo1/3 double knockout mice strain has a synergistic effect, inducing the formation and growth of GCTs. The loss of Foxo1/3/Pten contributes to the formation of GCTs because FOXO1/3 in granulosa cells regulates follicular development and apoptosis. This mouse model shows high expression of the Foxl2, Gata4, and Wnt4 genes, similar to what is observed in human GCT patients. Furthermore, the serum hormone profiles, which indicate elevated estradiol levels, high levels of activin (specially, βB) and inhibin, and low serum LH and FSH levels, suggest that the Foxo1/3 double-knockout mice can serve as a model for adult human GCTs. The tumor granulosa cells exhibit high expression of p-SMAD2/3 in the nuclei of granulosa cells, indicating that activin/TGFβ signaling is active in the formation of GCTs.
10. Oocyte-driven PIK3CA* mice
This mouse model was generated by crossing mice expressing oocyte-specific Cre-recombinase (GDF9-iCre) [69] with mice expressing constitutively active mutant PI3K (PIK3CA*) [70]. In these mice, the elevation of phosphatidylinositol (3,4,5)-trisphosphate levels within oocytes promotes the survival of follicles and anovulation due to endocrine abnormalities. This mouse model develops GCTs when the mice are mature, at 2 months of age [71]. The hormonal profiles show high levels of activin, inhibin, AMH, testosterone, and progesterone, and low levels of FSH and LH. The molecular signatures of this mouse model include high levels of SMAD3, FOXL2, and GATA4. This mouse might provide a good model for identifying the molecular mechanisms of GCT initiation and formation due to the absence of the mutation in the granulosa cells [7071].
Conclusion
Despite significant progress in understanding GCT biology, questions remain regarding the molecular mechanisms of JGCT and AGCT development. Although some mutations that induce the formation of AGCTs and JGCTs have been discovered, the events that initiate tumors and drive recurrence are still unclear. Therefore, suitable models are very important in understanding the molecular pathways underlying GCT formation. In particular, it is important to revisit these mouse models (Table 1) and reassess their characteristics compared to human GCTs based on histopathology, molecular pathways, and recurrence. Novel mouse models will be useful in answering challenging and persistent questions about GCT etiology. In conclusion, mouse models are powerful tools that aid in understanding the etiology and biological mechanisms driving the initiation and progression of GCTs, as well as help in the development of new detection methods and treatments.
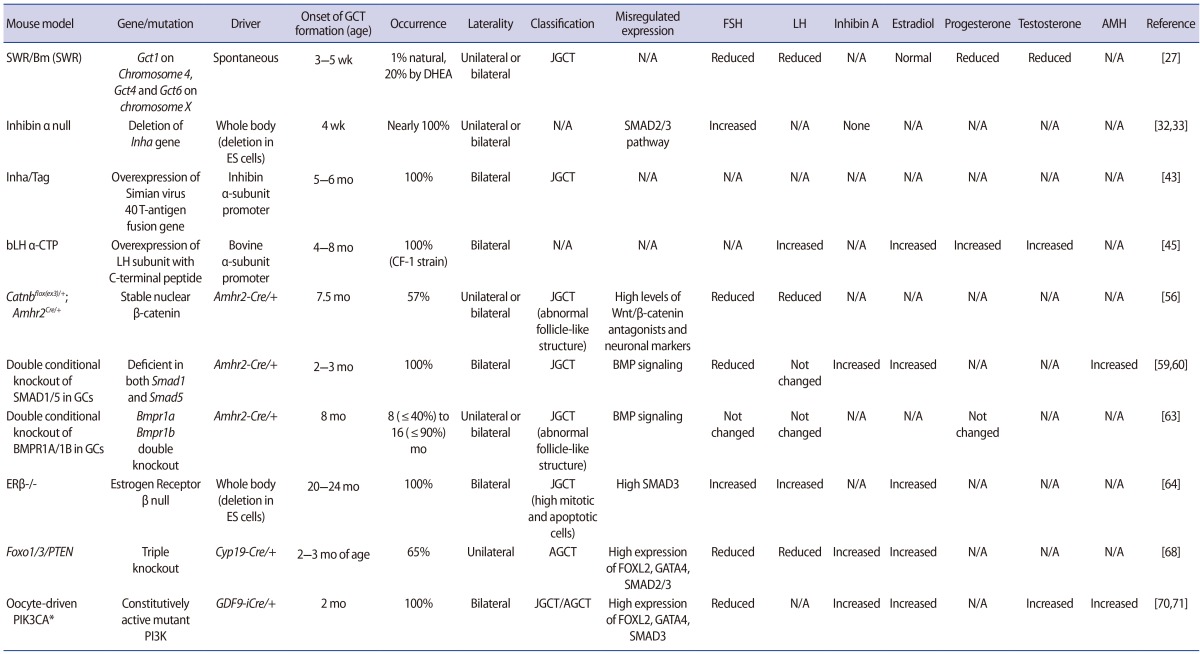
Currently proposed mouse models of granulosa cell tumors
Acknowledgments
I thank Dr. Marilia H. Cordeiro for her helpful suggestions and Stacey Tobin, Megan M. Romero, Alexandra S. Rashedi, and Maxwell E. Edmonds for editing this manuscript.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.