Mitochondria in reproduction
Article information

Abstract
In reproduction, mitochondria produce bioenergy, help to synthesize biomolecules, and support the ovaries, oogenesis, and preimplantation embryos, thereby facilitating healthy live births. However, the regulatory mechanism of mitochondria in oocytes and embryos during oogenesis and embryo development has not been clearly elucidated. The functional activity of mitochondria is crucial for determining the quality of oocytes and embryos; therefore, the underlying mechanism must be better understood. In this review, we summarize the specific role of mitochondria in reproduction in oocytes and embryos. We also briefly discuss the recovery of mitochondrial function in gametes and zygotes. First, we introduce the general characteristics of mitochondria in cells, including their roles in adenosine triphosphate and reactive oxygen species production, calcium homeostasis, and programmed cell death. Second, we present the unique characteristics of mitochondria in female reproduction, covering the bottleneck theory, mitochondrial shape, and mitochondrial metabolic pathways during oogenesis and preimplantation embryo development. Mitochondrial dysfunction is associated with ovarian aging, a diminished ovarian reserve, a poor ovarian response, and several reproduction problems in gametes and zygotes, such as aneuploidy and genetic disorders. Finally, we briefly describe which factors are involved in mitochondrial dysfunction and how mitochondrial function can be recovered in reproduction. We hope to provide a new viewpoint regarding factors that can overcome mitochondrial dysfunction in the field of reproductive medicine.
Introduction
Reproductive medicine aims to support healthy live births using assisted reproductive technology (ART). High-quality oocytes and fertilized embryos for transfer are required to achieve pregnancy [1]. The quality of oocytes and embryos is a major factor determining whether a pregnancy is successful. Unfortunately, there is no method to overcome ovarian dysfunction, oocyte aging, and poor embryo quality. In addition, there are several issues related to modern ART. For example, in vitro fertilization (IVF) patients tend to be relatively old and have a diminished ovarian reserve (DOR) and a poor ovarian response (POR) [2]. Many reproductive scientists have attempted to solve the major problems associated with ART and extensive research has sought to overcome aging [3]. However, these problems mostly remain unsolved and the mechanisms underlying DOR and POR remain unknown. Most of these problems are linked with mitochondrial dysfunction in the ovaries and during oogenesis. Therefore, the recovery of mitochondrial function may overcome aging, DOR, and POR. However, a strategy to overcome mitochondrial dysfunction has not been identified.
The fundamental properties of mitochondria must be better understood. Here, we summarize mitochondria, from their origin to their role in female reproduction. Mitochondria evolved via endosymbiotic processes—that is, the engulfment of bacteria by eukaryotic cells [4]. The evolution of mitochondria significantly enhanced energy production via aerobic metabolism in cells, leading to sexual reproduction rather than asexual reproduction. Aerobic metabolism by mitochondria has several benefits for cells, such as producing a large amount of bioenergy and supporting biomolecules. Mitochondrial functional activity is controlled by the nucleus (i.e., mitochondrial-nuclear communication) [5] and is important for maintaining mitochondrial function to facilitate cell proliferation, differentiation, and apoptosis.
Mitochondria have several unique characteristics in the reproductive system, such as their shape. Mitochondrial function is key for oogenesis in the ovaries [6], and mitochondrial dysfunction is associated with poor-quality oocytes and preimplantation embryos. Approaches to recover mitochondrial function in human oocytes and preimplantation embryos are being developed to overcome poor ovarian function (DOR and POR) and to improve the quality of oocytes and embryos. Unfortunately, there is currently no method of overcoming poor ovarian function or improving the quality of oocytes and embryos. Even the optimal culture conditions from the 2-pronuclear (PN) stage to the blastocyst stage remain unknown.
Several studies have reported that mitochondrial dysfunction is associated with oxidative stress caused by the byproducts of cellular metabolism, such as reactive oxygen species (ROS). Several factors associated with mitochondrial dysfunction, such as oxidative stress, may increase mutations of mitochondrial DNA (mtDNA) in aged oocytes; however, anti-ROS agents do not completely prevent the occurrence of mtDNA mutations caused by aging [7]. In particular, oxidative stress cannot explain the numerous age-related responses involved in mitochondrial dysfunction and the loss of activity in aged oocytes. Other factors seem to increase the number of dysfunctional mitochondria in aged oocytes. A deeper understanding of mitochondrial function and how it can be recovered is required to overcome aging, DOR, and POR in reproductive medicine.
This review aimed to summarize the origin and basic role of mitochondria in cells and their specific properties in reproduction. In addition, we present a new viewpoint regarding how mitochondrial function can be recovered in ovaries, oocytes, and embryos.
General roles of mitochondria in cells
1. The origin of mitochondria: endosymbiotic theory and eukaryotic origins
Two main competing theories have been reported about the origin of mitochondria. The evolution of mitochondria was a huge step forward, leading to enhanced cell properties due to the symbiotic relationship between the host cell and the endosymbiont [4]. The endosymbiotic theory reported by Lynn Margulis in 1967 is the most accepted theory. This theory states that mitochondria arose once during evolution and originated via endosymbiosis accompanied by gene transfer from the endosymbiont to the host [8]. Anaerobic mitochondria are puzzling for people who hold traditional views about the origins of mitochondria, but are explained by newer theories of mitochondrial evolution that were formulated to take into account the common ancestry of mitochondria and hydrogenosomes [9]. The presence of mitochondria in the common ancestor of eukaryotes continues to change the way we view the origin of eukaryotes, with endosymbiosis thought to play a more central role now than it did 20 years ago. The integral role of mitochondria in many aspects of eukaryote biology might reflect their role in the origin of eukaryotes.
Mitochondria in eukaryotes were inherited vertically from an archaebacteria-related host. The mitochondrial genome contains only 60 protein-coding genes, whereas the nuclear genome encodes more than 1,000 proteins that support mitochondrial function [10]. mtDNA constantly escapes from mitochondria and is integrated into nuclear DNA. Most mitochondrial proteins are encoded by nuclear genes, and many of these were endosymbiotically acquired from the mitochondrial ancestor [9]. Nuclear-mitochondrial communication impacts sexual reproduction processes, such as meiosis and the cell cycle. Mammalian reproduction is very advanced compared with the asexual reproduction of prokaryotic cells. Sexual reproduction is well organized to ensure that genes are inherited by the next generation, and that gene expression is precisely regulated. Therefore, the sexual reproduction of eukaryotic cells is complex and requires a large amount of bioenergy. The evolution of mitochondria dramatically enhanced adenosine triphosphate (ATP) production via processes including glycolysis and oxidative phosphorylation.
2. The role of mitochondria in production of bioenergy and synthesis of biomolecules
The major role of mitochondria, which are double membrane-bound subcellular compartments, is to supply eukaryotes with energy in the form of ATP and thereby meet their energy requirements. Mitochondria are the site of aerobic respiration, a complex biochemical process by which pyruvate is oxidized to CO2, generating reduced cofactors that drive the electron transport chain (ETC) to chemiosmotically fuel ATP synthesis. Mitochondria use oxygen as an electron acceptor in the final step of ATP production (Figure 1). Aerobic respiration supports eukaryotic cells and processes several important metabolites to support cell survival and bioenergy production (Figure 1) [10].

The general role of mitochondria and mitochondrial-nuclear communication. mRNA, messenger RNA; tRNA, transfer RNA; sRNA, small RNA; lnRNA, long non-coding RNA; CoA, coenzyme A; TCA, tricyclic antidepressant; ETC, electron transport chain; ADP,, adenosine diphosphate; ATP, adenosine triphosphate; SIRT, silent information regulator; HAT, histone acetyltransferase; AMPK, AMP-activated protein kinase.
Mitochondria catabolize nutrients for energy production, generate biosynthetic precursors of macromolecules, compartmentalize metabolites for the maintenance of redox homeostasis, and function as hubs for metabolic waste management. Mitochondrial metabolic pathways include several processes for biosynthesis of products such as nucleotides, fatty acids, cholesterol, amino acids, and glucose. Dysregulation of these pathways causes severe diseases such as cancer and aging [11].
The mitochondrial proteome typically contains more than 1,000 proteins that function in a wide variety of critical biochemical processes, including protein synthesis, amino acid and nucleotide metabolism, fatty acid catabolism, lipid, quinone, and steroid biosynthesis, iron-sulfur cluster biogenesis, apoptosis, and ion homeostasis [10].
3. Mitochondrial-nuclear communication
Mitochondrial-nuclear crosstalk regulates cellular functions such as differentiation and stress adaptation. Mitochondria supply metabolites required for epigenetic modifications and other nuclear activities, and mitochondrial proteins are found in the nucleus (Figure 1). Mitochondrial-nuclear communication coordinates several crucial cellular functions, including metabolic homeostasis, ROS generation, steroid synthesis, apoptosis, proliferation, differentiation, immunity, and aging [5].
Mitochondrial function and gene expression influence the nuclear epigenome via trafficking of metabolic enzymes and metabolites to the nucleus. The activities of epigenetic and epitranscriptomic enzymes are tightly regulated by the abundance and subcellular localization of metabolites [12]. RNAs and epigenetic enzymes translocate from the nucleus to mitochondria and regulate mitochondrial metabolism [12]. Mitochondrial RNA is modified by nuclear-encoded epitranscriptomic enzymes that regulate mitochondrial function.
Mitochondria have a circular genome, which is a remnant of their symbiotic bacterial origins. To coordinate complex cellular functions such as metabolism and stress responses, mitochondrial and nuclear genomes have evolved together [13]. The nuclear genome encodes more than 1,000 proteins required for mitochondria to function effectively [14]. However, the factors encoded in the mitochondrial genome that reside in the nucleus remain unknown. All 13 mtDNA-encoded proteins are structural components of the ETC and play a crucial role in oxidative phosphorylation [15]. A number of regulatory peptides encoded by the mitochondrial genome as short open reading frames have recently been identified [16]. Mitochondrial-derived peptides play diverse biological roles, including in stress responses and metabolic regulation, and contribute to mitochondrial communication. However, the mechanism by which these peptides regulate nuclear gene expression in response to cellular stress remains unclear.
4. The key role of mitochondria in cell aging and death and calcium homeostasis
Dysfunctional mitochondria are a major phenotype of aging and cell death; therefore, most mitochondrial studies are linked to aging. Anti-aging is the process of recovering or protecting mitochondrial function for rejuvenation. However, anti-aging is complex, and the underlying mechanism and contributing factors are unclear. Several types of programmed cell death have been reported, including apoptosis, necroptosis, and ferroptosis. Mitochondria also regulate calcium homeostasis by storing calcium ions depending on the cellular conditions. Therefore, mitochondria are important for the regulation of programmed cell death.
The ability of isolated mitochondria to take up Ca2+ from the cytoplasm and accumulate it in their matrix in an energy-dependent manner was first described as a key process for cell death and calcium homeostasis more than 40 years ago [17]. By the end of the 1970s, all the functional characteristics of the mitochondrial Ca2+ handling machinery had been described. Ca2+ was believed to be stored in mitochondria and mobilized upon cell activation. However, the demonstration that mitochondria in healthy living cells contain trace amounts of Ca2+ and the characterization of the mitochondrial Ca2 uniporter mentioned above (low influx rate at physiological Ca2+ concentrations in both resting and activated cells) convinced most investigators that mitochondrial Ca2+ uptake plays a marginal role in the overall control of Ca2+ homeostasis under physiological conditions and is important only in pathological situations. Necrotic cells are often characterized by electron-dense material comprising precipitates of Ca2+ phosphate, in mitochondria. The scientific community paid little attention to mitochondrial Ca2+ handling during the subsequent decade. However, some studies have indirectly suggested that mitochondria play an important role in Ca2+ handling in cell physiology [17].
Perturbations of mitochondrial Ca2+ handling have major pathophysiological consequences in several human diseases, including neurodegeneration. The expression of pro- and antiapoptotic genes modulates mitochondrial Ca2+ handling, with implications for cell survival and death [18].
Specific properties of mitochondria in reproduction
1. The bottleneck theory
Mitochondria contain DNA distinct from nuclear DNA. mtDNA is independently transmitted to progeny through germline development followed by fertilization. Heteroplasmic variants of mtDNA from primordial germ cells containing mutated mtDNA are segregated into primary and matured oocytes during meiotic division. In addition, maternal mtDNA is solely inherited because mitochondria of sperm are generally removed from the penetrated oocyte during fertilization. This restricted inheritance—the so-called mitochondrial genetic bottleneck—can lead to a random shift in the maternal-derived mtDNA mutational load between generations and consequently affect diverse pathogenic phenotypes in offspring along with mtDNA mutation levels (Figure 2).
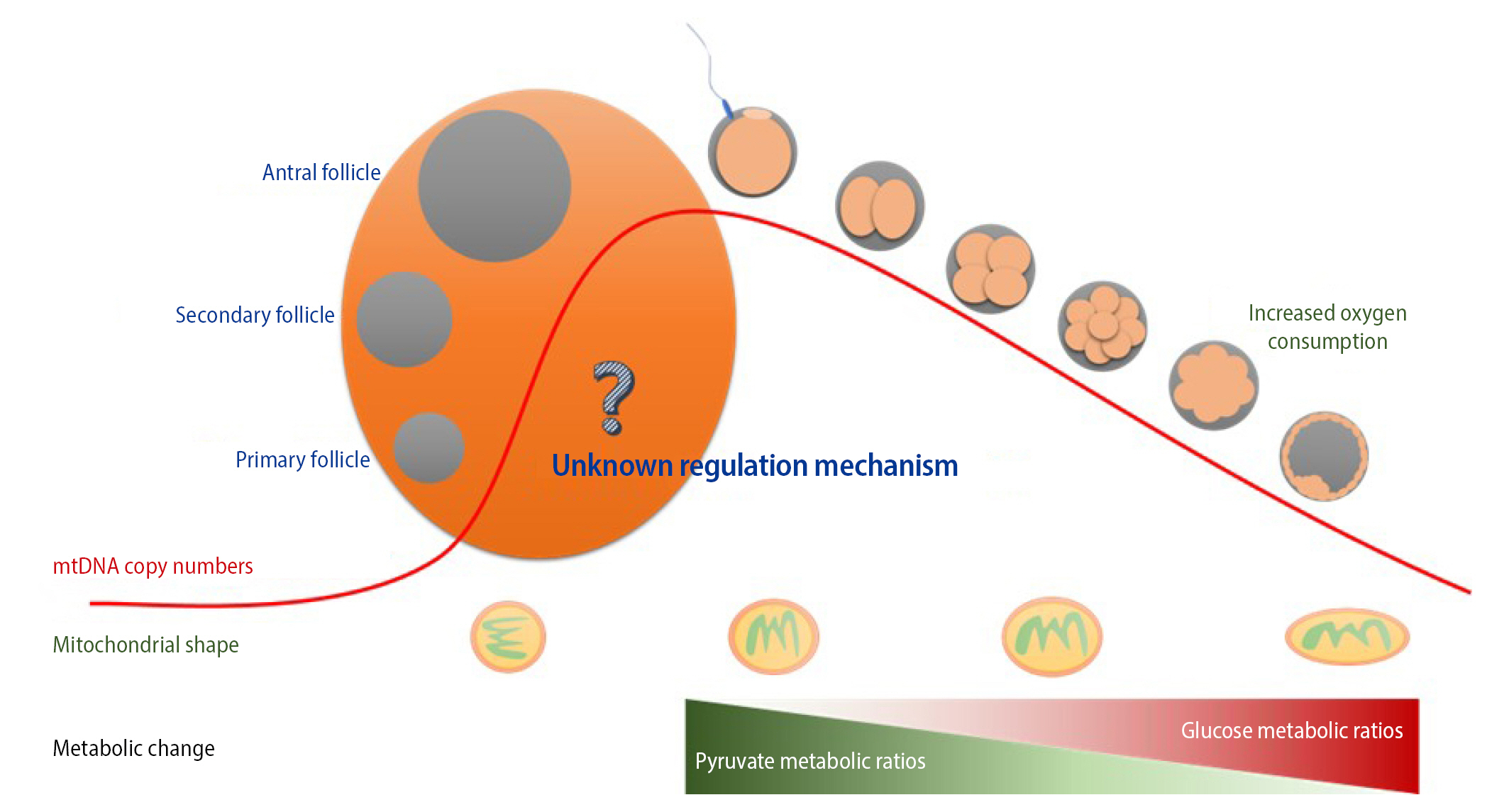
Characteristics of mitochondria in female reproduction. mtRNA, mitochondrial DNA.
This germline genetic bottleneck presumably involves two distinct phases: a passive reduction in mtDNA to an extremely low level, followed by an increase of mtDNA during fertilization and implantation [19-21]. These drastic quantitative changes in mtDNA probably occur at a very early stage of germ cell development [22], but the precise timing and regulatory mechanisms are unclear from the viewpoint of aging and reproduction [20,23,24]. Intriguingly, a recent study reported divergence in mtDNA between a mother and offspring that correlated with the mother’s age after childbirth due to the continued drift of heteroplasmy frequencies in oocytes in meiotic arrest [25]. Human phylogenetic analysis of 96 multigenerational families revealed that elevated de novo mutation frequencies in the mtDNA of female gametes with increasing maternal age intensified mother-child mtDNA divergence by the germline bottleneck effect. This implies that a child has a relatively high risk of inheriting a disease-causing mtDNA mutation from an asymptomatic, elderly, carrier mother and developing a disease. Despite extensive explorations of genetic mutations transmitted via mitochondria in mice and several other species, there is no strong evidence of a mitochondria selection mechanism during embryogenesis [25,26].
2. The role of mitochondria in ovaries and ovarian longevity
In infertile women, mitochondrial dysfunction is associated with ovarian aging. Mitochondrial properties are a significant factor in ovarian aging and longevity. The number and quality of oocytes decrease as women age, manifesting as DOR and POR, respectively [27]. Aberrations of mtDNA can be quantitative, such as changes in the mtDNA copy number and deletions, or qualitative, such as strand breaks, point mutations, and oxidative damage [28]. Mitochondrial damage may occur collaterally due to a primary disease or inflammatory process that overwhelms the mitochondrial capacity to eliminate oxidative byproducts [29]. This is likely to be the case for aging, obesity, diabetes, and other chronic inflammatory conditions. In other cases, mitochondrial dysfunction, such as that induced by congenital mutations of mitochondrial or nuclear DNA, is the primary cause of inflammation and premature aging [30]. In addition, other studies on reproduction have reported that factors such as mutation, oxidative stress, and strand breaks are associated with mtDNA dysfunction [31-33].
Therefore, it is imperative to define the mechanisms that lead to mitochondrial dysfunction and depletion because correction of these deficits will attenuate premature aging. In patients with premature ovarian aging, there is no strong evidence-based pharmacologic method to effectively rejuvenate mitochondria. However, several questions remain unanswered concerning mtDNA dysfunction and ovarian aging.
Mitochondrial dysfunction can be quantified by measuring mtDNA copies and deletions. To maintain mtDNA homeostasis, mitochondrial transcriptional factor A (TFAM), a histone-like protein, regulates transcription initiation and mtDNA copy number to ensure mitochondrial genome stability and protection [34]. TFAM may bind differently to mtDNA regions prone to oxidation and may regulate quantitative mtDNA dysfunction [35].
Reproductive aging is associated with a decreased mtDNA copy number. In humans, the mtDNA content of oocytes plays a crucial role in oocyte fertilization and embryo development [36]. The mtDNA content is significantly lower in oocytes of older women or women with DOR than in oocytes of younger women or women with a normal ovarian reserve [37]. Unfertilized oocytes of women with fertility problems also have a lower mtDNA copy number [38]. The positive correlation between oocyte mitochondrial mass and developmental competence indicates that pharmacologic approaches to increase the oocyte mitochondrial mass may be beneficial for fertility treatment.
mtDNA deletions are associated with ovarian aging. The increased frequency of a 4,977 bp deletion in postmenopausal women indicates that this deletion accumulates around the postmenopausal period [39]. Moreover, analysis of unfertilized MII oocytes retrieved from IVF patients showed that the mean age of patients harboring mtDNA deletions (38 years) was higher than that of patients without mtDNA deletions (31 years), suggesting that mtDNA deletions accumulate with increasing maternal age [40].
3. Mitochondrial shape in oocytes
There are more mitochondria in oocytes than in somatic cells, and mitochondria are smaller and rounder in the former than in the latter [41]. Moreover, the number and activity of mitochondria are tightly linked with oocyte quality owing to the key role of mitochondria in oocyte maturation [33]. The mitochondrial shape during oogenesis and embryo development is interesting (Figure 2). Mitochondria in primary oocytes have an almost circular shape and acquire an oval shape upon maturation [42]. The shape of mitochondria is related to their metabolic activity and replication in cells. However, the mechanism by which mitochondria change shape during oogenesis and embryo development remains unclear. The injection or transfer of mitochondria obtained from adult cells into oocytes is not a viable strategy because mitochondrial shape differs between them. Instead, the injection or transfer of mitochondria derived from oocytes or embryos is a better approach to improve the quality of oocytes and embryos [43]. The mitochondrial shape must be the same in the donor and recipient to overcome mitochondrial dysfunction in oocytes.
4. Changes in energy metabolism during preimplantation embryo development
Energy metabolism in cells is divided into glycolysis (anaerobic phase) and oxidative phosphorylation (aerobic respiration processes), both of which are observed during preimplantation embryo development from the one-cell stage to the blastocyst stage (Figure 2) [44]. In particular, numerous interactions between cytoskeletal proteins are actively involved in the regulation of energy metabolism, mitochondrial function, ATP production, and energy transfer during oocyte maturation and preimplantation embryo development [45-48].
In our previous study, we reported differences in mitochondrial movement and cytoskeletal integrity between young and old mouse oocytes [49]. The cytoskeleton exhibited rapid movement and optimal integrity in young oocytes compared with aged oocytes. Upon treatment with a microtubule disturber, actin cytoskeleton instability led to a significant decrease in mitochondrial motility and a low ATP production ratio in aged oocytes compared with young oocytes. Our data indicate that instability of the actin cytoskeleton is the primary cause of mitochondrial dysfunction in aged murine oocytes. These results suggest a relationship between cytoskeletal instability and mitochondrial dysfunction, which is related to mitochondrial dynamic properties, in oocytes of aged mice. Therefore, cytoskeletal stability may affect mitochondrial motility and bioenergy production during oocyte maturation and embryo development.
Recovery of mitochondrial function in aged ovaries and poor-quality oocytes and preimplantation embryos
1. Recovery of mitochondrial function in aged ovaries
Aged ovaries are characterized by inflammation, oxidative stress, apoptosis, necroptosis, and loss of calcium homeostasis [50-54]. These aging phenotypes are related to mitochondrial dysfunction. Current strategies to overcome ovarian aging include inhibiting apoptosis (e.g., with anti-ROS agents such as ascorbic acid, N-acetyl-L-cysteine, and melatonin), necroptosis, and inflammation and improving calcium homeostasis [55,56]. Some factors have been reported to protect and improve aged ovaries [56]. However, investigations on most of these factors remain at the research or pre-clinical level.
Necroptosis is a novel type of programmed cell death. It is regulated by molecules such as receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and receptor-interacting serine/threonine-protein kinase 3 (RIPK3), and plays roles in inflammation, survival, and disease. Phosphorylated mixed lineage kinase domain-like pseudokinase (MLKL) is the critical effector of necroptosis [57]. Necroptosis is induced by Toll-like receptors, death receptors, interferons, and other mediators. The results obtained using a mouse model reveal that the deregulation of necroptosis is associated with pathological conditions such as cancer, neurodegenerative diseases, and inflammatory diseases. Recent studies reported that RIPK3 also directly activates the formation of the inflammasome in response to cellular stress or microbial infection to activate caspase-1 and caspase-11. However, it is unknown how RIPK3 activates NOD-like receptor pyrin domain-containing 3 (NLPR3)-mediated inflammasome formation with or without the involvement of MLKL. Some models propose that RIPK3 acts as a scaffold to recruit a complex containing RIPK1, FAS-associated protein with death domain (FADD), and caspase-8 [58-61]. Upon formation of this complex, caspase-8 promotes the maturation of naive interleukin-1β by an unknown mechanism or activates caspase-1 in the NLPR3 inflammasome. However, caspase-8 also prevents RIPK3-MLKL-mediated assembly of the NLPR3 inflammasome [57]. Several studies have suggested that necroptosis, a regulated form of necrosis, has similar morphological features to those of necrosis and occurs in human ovaries [62]. Stress may mediate necroptosis in mammalian ovaries. Inflammation is elevated in aging ovaries, but it is unclear whether this is related to necroptosis. An anti-necroptosis factor may be able to overcome ovarian aging; however, this has not been investigated.
Inhibition of necroptosis has been investigated as a way to protect against necrosis and reduce inflammation in the ovaries [53]. The inhibition of necroptosis enhances the recovery of primordial follicles and development of antral follicles in aged ovaries. Oct3/4, Nanog, and p63 are markers of primordial follicles that regulate the proliferation of stromal cells and differentiation of granulosa cells during the development of preantral follicles [63]. The inhibition of necroptosis may facilitate controlled ovulation stimulation by upregulating Oct3/4 and Nanog, thereby enhancing the survival of primordial follicles in aged ovaries. p63 is a regulator of meiosis and is important for cell cycle control in primordial follicles [64]. The inhibition of necroptosis promotes antral follicular development in aged ovaries. It also increases mitochondrial respiration ratios in aged human cumulus cells compared with young human cumulus cells. Prostaglandins regulate both the gonadotrophin-independent and -dependent phases in the ovary. In summary, an anti-necroptosis agent promotes follicular development by enhancing mitochondrial function and steroidogenesis, reducing inflammation, and thereby recovering ovarian function.
2. Cytoplasmic injection into oocytes and PN transfer
Aging oocytes are characterized by decreased mitochondrial function and accumulated mtDNA mutations. Potential ways to avoid the transmission of incurable mitochondrial diseases have been investigated in terms of ART. Several strategies related to mitochondrial replacement therapy (MRT) have been suggested and discussed for clinical applications. The first technique, cytoplasmic transfer, was devised to overcome infertility. It involves transferring approximately 5% to 15% of the cytoplasm from a healthy donor to the recipient oocyte of a patient with mutated mtDNA via intracytoplasmic sperm injection (ICSI). The reconstituted zygote contains the original parental nuclear DNA and a mixture of mtDNA from the recipient and donor oocytes [21,65-68]. The donor ooplasm is thought to contain healthy mitochondria and other beneficial cytosolic factors that promote development and embryogenesis [41,69,70]. Although this technique was initially developed to improve oocytes with poor-quality cytoplasm using animal models, it has been employed as an ART and led to clinical outcomes [21,66]. However, it is not used as a primary clinical technique because of the possibility of heteroplasmy and chromosomal abnormalities in the derived embryos [71,72].
PN transfer, which involves transferring the PNs of a zygote with abnormal mtDNA to another zygote with healthy mtDNA, was developed as an MRT. This method requires both recipient and donor oocytes to be fertilized by the intended partner’s sperm via ICSI or IVF. After PNs have formed in both zygotes, PNs from the defective oocyte are transferred into the enucleated zygote with normal mtDNA via micromanipulation, leading to reconstruction of a zygote containing nuclear DNA of the original parents and healthy mtDNA [66,72]. The first human study of PN transfer was published by Craven et al. [73] using abnormal human zygotes (one or three PNs). It reported that the level of mutant mtDNA was reduced to 2% or was undetectable in reconstructed zygotes after cleavage and subsequent blastocyst development in vitro. A pregnancy was established using reconstructed zygotes after PN transfer and a fetus developed without heteroplasmy and with a normal karyotype, although it did not develop to term [74]. As a new PN transfer protocol, PN transplantation soon after the completion of meiosis improves the survival rate of reconstructed zygotes by minimizing mtDNA transport during the oocyte vitrification procedure [75]. PN transfer has started to be clinically applied and has been explored in several species, including humans. However, this method must be further optimized, and its usefulness must be proven in many more clinical cases to ensure that it can be reliably used to avoid mitochondrial disease or as a speculative treatment for infertility.
3. Anti-ROS agents to protect against ovarian aging
Mitochondrial dysfunction is related to ROS, which cause mtDNA mutation and protein damage and thereby alter metabolic regulation. Most aging phenotypes are related to mitochondrial dysfunction. Therefore, scientists have sought to elicit anti-aging effects and treat aging-related diseases by recovering mitochondrial function. Chemical-based materials such as anti-ROS agents (e.g., nicotinamide adenine dinucleotide [NAD]-related materials) have been investigated to overcome mitochondrial dysfunction [76-78]. Anti-ROS products have applications to protect against mtDNA damage and elicit anti-aging effects. However, a study of human oocytes reported that an anti-ROS agent could not overcome oocyte aging. Other potential factors must be studied and further evidence must be obtained regarding mitochondrial dysfunction in aging.
The functional activity of mitochondria is associated with the quality of oocytes and embryos. Therefore, the recovery of mitochondrial function is very important to overcome oocyte aging and improve embryo quality. No reagent that can recover mitochondrial function in aged oocytes and embryos has been reported. Such a reagent may be very useful to overcome aging during female reproduction.
Several studies have reported the effects of anti-ROS agents as reagents to restore mitochondrial function during the in vitro maturation of aged oocytes and development of embryos derived from these oocytes. Mitochondrial function is perturbed in aged oocytes and embryos due to ROS-induced damage are byproducts of oxygen metabolism at the inner mitochondrial membrane. Mitochondrial bioenergy production is regulated by components of the ETC such as NAD, flavin adenine dinucleotide (FAD), and cytochrome B, C, and A in the inner mitochondrial membrane, and ATP is synthesized as bioenergy by ATP synthase. NAD and NAD phosphate are common mediators of various biological processes such as energy metabolism, mitochondrial function, calcium homeostasis, oxidative stress, gene expression, immunological functions, aging, and cell death. However, NAD is a major ROS producer during metabolic processing in the ETC. Under normal metabolic conditions, complex III is the main site of ROS production. More ROS are produced during oogenesis, maturation, and the development of aged oocytes and embryos. Optimal levels of ROS are important for cellular processes such as autophagy activation. However, high levels of ROS are associated with damage to mtDNA coding regions. The increased sensitivity of mtDNA to oxidative damage has led to the concept of a vicious cycle, in which an initial ROS-induced impairment of mitochondria leads to increased oxidant production, which in turn leads to further mitochondrial damage [79]. Old mitochondria appear morphologically and functionally altered and produce more oxidants and less ATP than young mitochondria.
Anti-ROS agents improve the in vitro maturation ratios of aged oocytes by reducing ROS-induced damage and inner mitochondrial membrane protein damage. Consequently, these agents recover mitochondrial function in aged mouse embryos. Anti-ROS agents also improve the development of old embryos compared with no treatment. The hatched ratio at the blastocyst stage was 10% higher for old embryos treated with anti-ROS agents than for control old embryos [80].
Bioenergy production by mitochondria is very important for chromosome segregation during oocyte maturation and embryo development [81,82]. Upon mutation and deletion of mtDNA, mitochondria cannot produce the required energy for chromosome segregation in the germinal vesicle breakdown (GVBD), MI, and MII stages. Anti-ROS agents can be added to the culture media of embryos in order to reduce ROS-induced damage, thereby significantly improving mitochondrial function and ATP production. Supplementation of a reagent to restore mitochondrial function might improve the quality of oocytes and embryos, and subsequently lead to healthy pregnancies in older women. We believe that large, properly randomized and controlled trials are needed to test the effect of mitochondrial function recovery reagents on clinical reproductive outcomes.
4. New strategies to recover mitochondrial function in oocytes and embryos
Maternal age affects the actin cytoskeleton and is associated with several novel criteria for human oocyte quality [83]. The functional activity of mitochondria is crucial for chromosome segregation and oocyte quality and viability. Older women have lower pregnancy rates and a higher chance of producing embryos with chromosomal abnormalities than younger women, and aneuploidy ratios are significantly increased in aged gametes and zygotes [84]. There is no reverse process for zygote aging, and no therapeutic tools are available for aged zygotes. In addition, dysfunctional mitochondria are key elements in aged zygotes. Mitochondrial function cannot be restored to overcome or reverse aging.
We found an association between cytoskeletal instability and mitochondrial functional activity, which is related to mitochondrial dynamic properties, in oocytes of aged mice [49]. According to these results, mitochondrial properties, including migration and proliferation, are regulated by microtubule stability, contrary to commonly held views regarding mitochondrial bioenergy production in mainstream biology. Indeed, microtubule integrity dramatically differs between young and old oocytes (Figure 3). Infertility in older women may be overcome by treatments that improve mitochondrial function in aged oocytes and embryos.
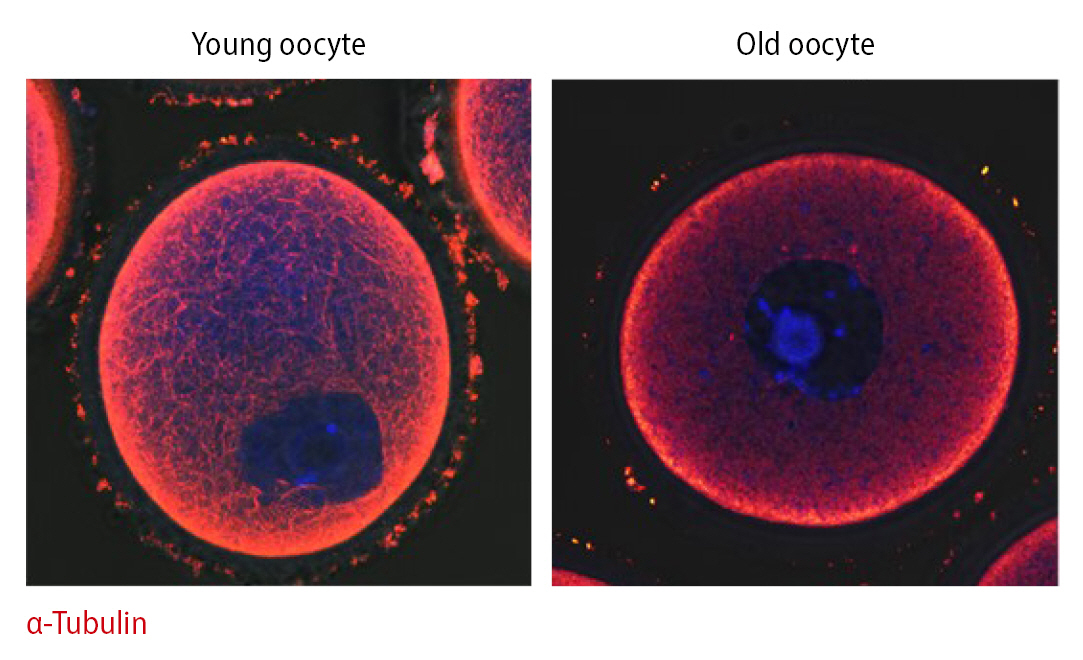
Immunofluorescence staining of α-tubulin (red) and DNA (Hoechst 33342, blue) in germinal vesicle oocytes of young (10 weeks old) and aged (55 weeks old) mice.
Depending on age, mitochondrial motility along the actin cytoskeleton may play an important role in oocyte quality [49]. Aged oocytes mature slower than young oocytes. During GVBD, dynamic mitochondrial motility was higher in young oocytes than in older oocytes. Due to increased mitochondrial densities in nuclear areas during the MI stage in young oocytes, mitochondrial location varied significantly between the maturation stages and groups. By contrast, mitochondrial motility was low in aged oocytes, and mitochondria did not preferentially accumulate in nuclear areas of these oocytes during maturation. Based on these observations, changes in mitochondrial location are dependent on the stage of aging, in agreement with a previous study. This phenomenon may be linked to the stability of the actin cytoskeleton during embryo development, and increased stability of the actin cytoskeleton results in aneuploidy in embryos.
Conclusion
Mitochondrial function is a major regulatory element to overcome ovarian aging and improve the quality of oocytes and embryos. Here, we presented the relationship between mitochondria and gamete quality. However, the major mechanism regulating mitochondrial function in reproduction has not been completely elucidated. The enhancement of mitochondrial function is very useful in ART. An activator of mitochondrial function will restore the competence of oocytes and preimplantation embryo development. Supplementation of culture media with an enhancer of mitochondrial function is useful to improve the quality of oocytes and preimplantation embryos. The recovery of mitochondrial function will promote healthy pregnancies in patients with POR or DOR.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Author contributions
Conceptualization: JHL. Visualization: YJK. Writing-original draft: MHK, JHL. Writing-review & editing: MHK, JHL.