A healthy delivery of twins by assisted reproduction followed by preimplantation genetic screening in a woman with X-linked dominant incontinentia pigmenti
Article information
Abstract
The purpose of this study is to report a successful twin pregnancy and delivery in a female patient with X-linked dominant incontinentia pigmenti (IP) who underwent assisted reproductive technology followed by preimplantation genetic screening (PGS). A 29-year-old female with IP had a previous history of recurrent spontaneous abortion. A molecular analysis revealed the patient had a de novo mutation, 1308_1309insCCCCTTG(p.Ala438ProfsTer26), in the inhibitor of the kappa B kinase gamma gene located in the Xq28 region. IVF/ICSI and PGS was performed, in which male embryos were sexed using array-based comparative genomic hybridization (aCGH). After IVF/ICSI and PGS using aCGH on seven embryos, two euploid male blastocysts were transferred with a 50% probability of a viable male pregnancy. The dizygotic twin pregnancy was confirmed and the amniocentesis results of each twin were normal with regard to the mutation found in the mother. The patient delivered healthy twin babies during the 37th week of gestation. This case shows the beneficial role of PGS in achieving a successful pregnancy through euploid male embryo gender selection in a woman with X-linked dominant IP with a history of multiple male miscarriages.
Introduction
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger disease, is a rare X-linked dominant genodermatosis that affects varying parts of the body, including the dermatological, ocular, nervous, and immune systems, and is generally lethal in male fetuses [1,2]. IP is caused by mutations in the inhibitor of kappa B kinase gamma (IKBKG) gene, also known as the nuclear factor-kappa B essential modulator gene, in the Xq28 region. The most common mutations (60%-80% of cases) involve a large-scale deletion of IKBKG exons 4 through 10 [3,4,5].
Though IP is related to recurrent spontaneous abortions in male fetal pregnancies, there have been few reports of attempts to achieve successful pregnancies in patients with this condition through assisted reproductive technology and genetic testing techniques. In the past, patients with IP underwent in vitro fertilization and male embryo selection using preimplantation genetic diagnosis (PGD) with fluorescent in situ hybridization (FISH), resulting in either a normal male pregnancy or early miscarriage in the case of affected male fetuses while excluding possible female carriers [6,7,8]. However, no cases of successful childbirth resulting from this approach have yet been reported. Recent case reports of PGD in patients with IP have included molecular analyses of the IKBKG gene through polar body biopsies, but this method does not involve a comprehensive genomic analysis [9,10].
We report a successful pregnancy and delivery in a female patient with IP who underwent in vitro fertilization/intracytoplasmic sperm injection and preimplantation genetic screening (PGS) using array-based comparative genomic hybridization (aCGH) followed by a prenatal diagnosis.
Case report
A 29-year-old patient with known IP was referred to Fertility Center of CHA Gangnam Medical Center for fertility therapy. She had manifested skin erythema followed by vesicles as a neonate. Patchy hyperpigmented skin lesions subsequently appeared in multiple sites. At the time of her first visit to our institution, she had only mild skin lesions involving scattered hyperpigmented spots in the abdomen and upper legs. She had experienced three early spontaneous pregnancy losses during three years of marriage. No anatomical, immunological, thrombophilic or endocrinological factors contributed to recurrent spontaneous abortions in this patient. She had been clinically diagnosed with IP by Landy and Donnai's criteria [11] and by skin biopsy at ten years of age in the dermatology department of the referring hospital, with no family history. A cytogenetic analysis of this couple revealed normal karyotypes, but confirmatory molecular genetic analysis of IP had not been completed when assisted reproductive technology was applied. After genetic counseling, she was scheduled for an IVF cycle with PGS using aCGH to select euploid male embryos. Simultaneously, mutation screening was performed using polymerase chain reaction-direct sequencing to identify her pathogenic mutation, which is crucial for further genetic counseling and prenatal diagnosis. Genomic DNA was extracted from a blood sample. All exons and intron boundaries of the IKBKG gene were analyzed and a pathogenic mutation was identified in IKBKG exon 9 (1308_1309insCCCCTTG(p.Ala438ProfsTer26)). A seven-base insertion of CCCCTTG at position c.1309 was identified, which had resulted in a frameshift in which the 438th amino acid was changed from alanine to proline and a premature stop codon occurred at the 464th codon (Figure 1).

Polymerase chain reaction-direct sequencing in the IKBKG gene of the patient: a pathogenic mutation is present in IKBKG exon 9(1308_1309insCCCCTTG(p.Ala438ProfsTer26)), in which the seven-base sequence CCCCTTG was inserted at position c.1309, resulting in a frameshift (the 438th amino acid changed from alanine to proline) and a premature stop codon at the 464th codon. NEMO, nuclear factor-kappa B (NF-κB) essential modulator; IKBKG, inhibitor of κB kinase gamma; PB1, first polar body.
A total of nine oocytes were retrieved after controlled ovarian hyperstimulation with gonadotrophin-releasing hormone antagonist protocol (cetrorelix, Cetrotide, Merck Serono Europe Ltd., London, UK; 0.25 mg) using recombinant follicle-stimulating hormone (recombinant follitropin alfa; GONAL-f, Merck Serono S.p.A, Modugno, Italy, 225 IU daily), of which seven were fertilized using the intracytoplasmic sperm injection procedure. In each of the seven eight-cell stage embryos, a single blastomere was biopsied on day three and subjected to aCGH analysis by a commercial laboratory (MGMED Co., Seoul, Korea). The laboratory reported that there were two euploid male embryos, one euploid female embryo, and four aneuploid male embryos (-16, -8/+15, +20, +2/+3/+6/+11/-20) (Figure 2). Both of the euploid male embryos, which were grade two mid-blastocysts, were transferred on day five. After 12 days, serum beta human chorionic gonadotropin was 442.17 mIU/mL, increasing to 4,676 mIU/mL six days later. A clinical pregnancy was confirmed with two gestational sacs and twin fetuses showing viable heartbeats by ultrasonography at the sixth week of gestation. The patient underwent amniocentesis for a confirmatory prenatal diagnosis during the seventeenth week of pregnancy and both fetuses showed normal male karyotypes. They were shown to be normal for the mutation found in the mother through polymerase chain reaction-direct sequencing of the cultured amniocytes. This patient delivered healthy live twin babies during the 37th week of gestation with no obstetric or neonatal complications.
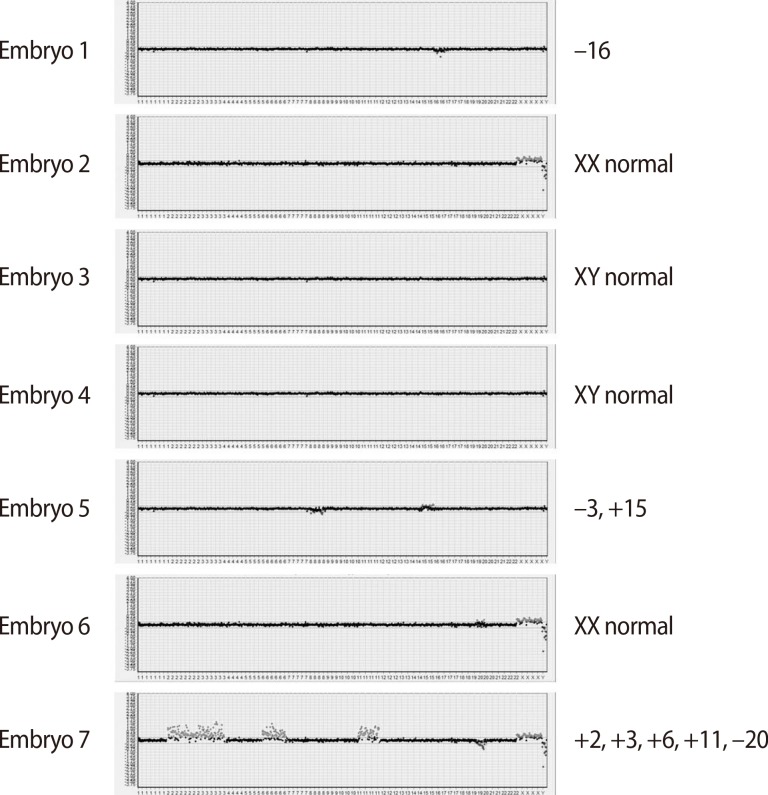
Array-based comparative genomic hybridization results of the seven embryos.
Discussion
IP is a rare X-linked dominant genodermatosis with its incidence of 0.7-2/100,000 newborns [12,13,14], which is lethal in males in utero in 97% of cases [15]. As a result, many women with IP have recurrent early miscarriages [16]. Although IP is usually lethal in males, approximately 72 cases of surviving male fetuses with IP have so far been reported [17]. The IP Consortium has proposed three mechanisms resulting in the survival of males carrying a mutation in the IKBKG gene: hypomorphic alleles, the 47,XXY karyotype (Klinefelter syndrome), and somatic mosaicism [16]. IP presents multisystemically but especially dermatologically, involving four typical stages of vesiculo-bullous, verrucous, hyperpigmented, and hypopigmented skin. Diagnostic criteria for IP have traditionally been based on the clinical features established by Landy and Donnai [11]. Several researchers have recently proposed that these criteria should be updated to reflect a firmer molecular understanding of how the nuclear factor-κB pathway is affected by the IKBKG gene mutation [18,19]. Approximately 65% of those mutations occur de novo and 69 different mutations have been reported [5,20,21].
The patient in this study had a phenotypically very mild form of IP that only manifested dermatologically. Her family history, including her parents and one brother, showed no evidence of IP. She had a de novo insertion/deletion (indel) mutation causing a frameshift and a premature stop codon, which occurs in 10% of IP cases. Her mutation in IKBKG exon 9, 1308_1309insCCCCTTG(p.Ala438ProfsTer26), is the first reported genetic aberration affecting the IKBKG gene. This finding is analogous to other mutations, which have also been reported just once [21].
Many cases of recurrent spontaneous abortions in patients with IP have been reported, reflecting its X-linked dominant nature and lethality to male fetuses, resulting in the suggestion that PGD should be used for patients with IP [22]. However, only a few cases have been reported of PGD used in embryos from IP-affected females in infertile couples who underwent IVF procedures (Table 1) [6,8,9,10]. FISH methods have conventionally been used to sex male embryos in the course of PGD in patients with IP, which entails the possibility of either viable male fetal pregnancies or miscarriages if affected male embryos are transferred [6,8]. However, in all of these case reports using FISH methods with fewer than six chromosome probes, the fetuses were spontaneously aborted before confirmatory prenatal diagnosis resulting from chromosomal aneuploidies which were not tested.
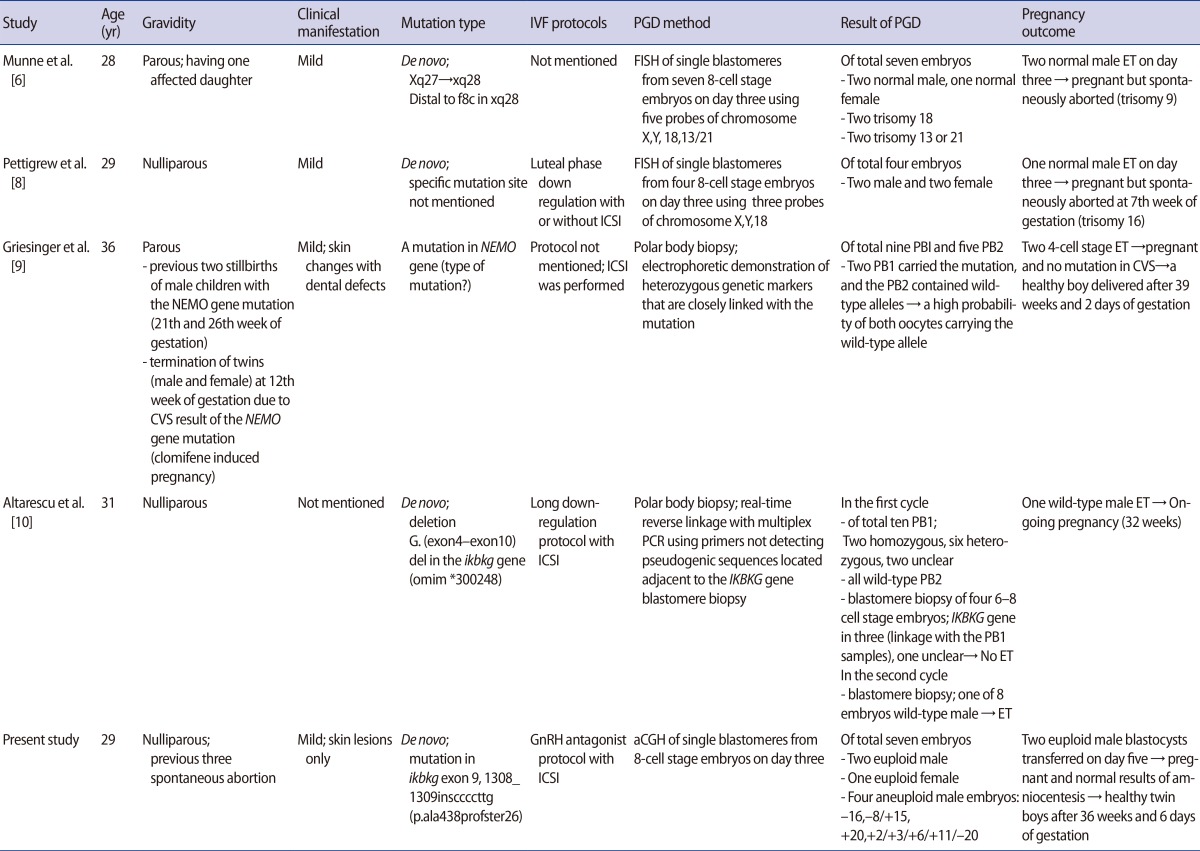
Summary of published PGD cases in patients with incontinentia pigmenti
In two recent case reports, the PGD for patients with IP was performed through polar body analysis strategies using polymerase chain reaction primers of the IKBKG gene to focus on the molecular basis of IP. The authors reported the delivery of a healthy boy and an ongoing pregnancy with one male fetus after 32 weeks of gestation [9,10]. In a more recent study evaluating 151 PGD cycles using polar body analysis for de novo mutations in 38 different genetic disorders, the researchers applied eight PGD cycles to five patients with IP. They reported four births from eight embryos, which were transferred after sequential polar body analysis and embryo karyotyping of blastomeres for several chromosomes by polymerase chain reaction [23].
In this case, we also selected male embryos with a 50% probability of a viable male pregnancy. However, we performed PGS as part of the embryo transfer process followed by amniocentesis with a molecular analysis of possible IP after the clinical confirmation of pregnancy. The patient delivered healthy dizygotic twin babies at the 37th week of gestation. We were able to prevent a miscarriage arising from an aneuploid pregnancy involving other autosomal mutations through a whole chromosome analysis using aCGH instead of only sexing the embryo, which was a significant difference from previous studies.
Although successful pregnancy outcomes have been presented in recent case reports in which polar body analysis was used, these reports have mainly focused on the molecular analysis of possible mutations inherited from the mother, without performing whole genome analysis. It is well known that aneuploidies are common in early human embryos [24,25]. According to a study that used FISH probes for chromosomes X, Y, 13, 15, 16, 17, 18, 21, and 22 in 6054 cleavage-stage embryos [26], 70% of embryos showed chromosomal abnormalities. In another study analyzing aCGH of 70 single blastomeres, the authors reported that 55.7% of the blastomeres were diploid, 44.3% contained chromosomal abnormalities, and 29% were abnormal cells with structural aberrations [25]. The relatively high aneuploidy rate of 57% previously reported in IP cases [6] corresponds to the aneuploidy found in four of the seven embryos in our case. In light of this, embryo selection in IP patients through comprehensive genomic analytical techniques like aCGH, as performed in this study, is a promising approach.
Recently, next-generation sequencing has emerged as a PGS strategy both for mutation target diagnosis and simultaneous whole genome analysis [27,28]. Another very promising study has been published about single-gene disorders like IP, but the clinical applicability of the relevant technology remains to be confirmed [29].
In conclusion, this case shows the beneficial role of PGS in achieving successful pregnancy through sexing euploid embryos in a woman with X-linked dominant IP who had experienced multiple male miscarriages.
Notes
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2009-0093821).
No potential conflict of interest relevant to this article was reported.