Influence of hydrogel encapsulation during cryopreservation of ovarian tissues and impact of post-thawing in vitro culture systems in a research animal model
Article information
Abstract
Objective
Using domestic cats as a biomedical research model for fertility preservation, the present study aimed to characterize the influences of ovarian tissue encapsulation in biodegradable hydrogel matrix (fibrinogen/thrombin) on resilience to cryopreservation, and static versus non-static culture systems following ovarian tissue encapsulation and cryopreservation on follicle quality.
Methods
In experiment I, ovarian tissues (n=21 animals; 567 ovarian fragments) were assigned to controls or hydrogel encapsulation with 5 or 10 mg/mL fibrinogen (5 or 10 FG). Following cryopreservation (slow freezing or vitrification), follicle viability, morphology, density, and key protein phosphorylation were assessed. In experiment II (based on the findings from experiment I), ovarian tissues (n=10 animals; 270 ovarian fragments) were encapsulated with 10 FG, cryopreserved, and in vitro cultured under static or non-static systems for 7 days followed by similar follicle quality assessments.
Results
In experiment I, the combination of 10 FG encapsulation/slow freezing led to greater post-thawed follicle quality than in the control group, as shown by follicle viability (66.9%±2.2% vs. 61.5%±3.1%), normal follicle morphology (62.2%±2.1% vs. 55.2%±3.5%), and the relative band intensity of vascular endothelial growth factor protein phosphorylation (0.58±0.06 vs. 0.42±0.09). Experiment II demonstrated that hydrogel encapsulation promoted follicle survival and maintenance of follicle development regardless of the culture system when compared to fresh controls.
Conclusion
These results provide a better understanding of the role of hydrogel encapsulation and culture systems in ovarian tissue cryopreservation and follicle quality outcomes using an animal model, paving the way for optimized approaches to human fertility preservation.
Introduction
The survival rate of cancer patients has dramatically increased due to tremendous progress in diagnosis and medical treatments. Nonetheless, female cancer patients in prepubescence or reproductive age who have undergone gonadotoxic anticancer treatments, such as alkylating chemotherapy or ionizing radiotherapy in the pelvic or abdominal regions or total body radiation, may have a high risk of fertility impairment [1]. Over recent decades, ovarian tissue cryopreservation and transplantation have been introduced as adequate options for female fertility preservation and restoration after patients recover from cancer [2]. So far, these technologies have resulted in more than 130 clinical cases of live births worldwide [3]. However, the risk of malignant cell reimplantation after ovarian tissue transplantation is a serious concern and should be addressed, especially in patients with leukemia [3]. In order to ensure the safety of fertility restoration without reseeding of cancer cells, the newly developed promising strategies of in vitro follicle activation (IVA) and in vitro follicle growth (IVG) have been extensively attempted as sequentially imperative steps prior to the construction of a cancer-free microenvironment for isolated follicle development [1].
Ideally, the in vitro activation of primordial follicle development to the pre-antral or secondary follicle stage mainly involves culturing cryopreserved cortical ovarian tissues [1,2,4]. Ovarian tissues are routinely cryopreserved by slow-freezing or vitrification methods using optimized freezing media and protocols that differ among laboratories [5]. Recently, a novel tissue engineering technology using biodegradable hydrogels or polymers (e.g., alginate, fibrin-collagen, fibrin-thrombin clot or three-dimensional (3D)-printed microporous hydrogel scaffolds) was introduced in reproductive medicine research, involving the encapsulation of immature testicular tissue fragments or prototype construction of scaffolds for follicle culture using fibrin-thrombin in various concentrations [4,6]. The porosity of this biodegradable matrix allows angiogenic factors and other essential nutrient components, such as epidermal growth factor and vascular endothelial growth factor (VEGF) [1,4], to be delivered into the gel network. Angiogenic factors and their protein phosphorylation (e.g., protein kinase B [Akt] and tyrosine kinase I [Tie1]) have been considered crucial keys for cell survival, graft neovascularization, and long-term in vitro tissue culture [6-8].
To date, very few tissue engineering technology studies related to ovarian tissue cryopreservation and in vitro culture have been conducted. The fundamental in vitro tissue culture protocol (static culture system) with sequential culture media changed every 1–2 days has been generally performed [1]. However, this culture system lacks medium circulation mimicking natural vascularization, which diminishes cell-to-cell interactions [9]. To address these issues, non-static culture systems using engineered fluid flow manipulation through the fine control of micro-fluid volume (known as microfluidics) have been actively explored in reproductive medicine laboratories [9,10]. A previous study in a mouse model demonstrated that a non-static culture system could potentially support follicle growth and granulosa cell luteinization after long-term culture for 14 days [9]. Although various attempts of ovarian tissue cryopreservation and transplantation have been successful, studies on ovarian tissue cryopreservation together with primordial follicle activation prior to IVG remain limited.
Due to defects in homologous genes resulting in similar molecular, pathological, and clinical phenotypes as in humans [11], domestic cats have been extensively studied as experimental models in a variety of research fields including genetic disorders, viral neuropathology, antiretroviral therapy, allergic asthma, type 2 diabetes mellitus, and assisted reproductive technology [12,13]. Thus, our goal was to address the lack of information on ovarian tissue cryopreservation in hydrogels and IVG using domestic cats as a biomedical research model. The main objectives of the present study were to characterize the influence of ovarian tissue encapsulation in a biodegradable matrix (with different concentrations of fibrinogen) on cryopreservation outcomes, and the influence of static versus non-static culture systems following encapsulation and cryopreservation on ovarian tissue integrity and follicle quality.
Methods
1. Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and all antibodies from Cell Signaling Technology (Danvers, MA, USA), unless otherwise indicated.
2. Experimental design
1) Experiment I: influence of ovarian tissue encapsulation on cryopreservation outcomes
A total of 27 ovarian cortical pieces (0.2 cm×0.2 cm×0.1 cm; width×length×thickness) from each animal (n=21; total of ovarian fragments=567) were retrieved and randomly allocated to different groups; fresh controls, encapsulation without or with 5 mg/mL fibrinogen, 50 IU/mL thrombin, and 0.1 ng/mL VEGF (5 FG) or encapsulation without or with 10 mg/mL fibrinogen, 50 IU/mL thrombin, and 0.1 ng/mL VEGF (10 FG) (Figure 1A). Ovarian cortical pieces from each group were then randomly selected for further equilibration and cryopreservation procedures (either slow freezing or vitrification) and evaluated for follicle viability (neutral red staining), normal morphology (histology and Hoechst 33342), follicle density (the number of follicles with normal morphology per 1 mm3; histology). The evaluation of Tie1 and VEGF receptor (VEGF-R) phosphorylation (Western blot analysis) was performed after cryopreservation only.

Designs of experiments I (A) and II (B). 5 FG, 5 mg/mL fibrinogen, 50 IU/mL thrombin, and 0.1 ng/mL VEGF; 10 FG, 10 mg/mL fibrinogen, 50 IU/mL thrombin, and 0.1 ng/mL VEGF; VEGF, vascular endothelial growth factor.
2) Experiment II: influence of static and non-static culture systems following encapsulation and cryopreservation on ovarian tissue integrity and survival
The design of experiment II was based on the findings from experiment I (Figure 1B). A total of 10 animals were included. ovarian cortical tissues (27 pieces per animal; total of ovarian tissue fragments=270) were divided into three major groups; fresh control, slow freezing after encapsulation in 10 FG, or vitrification after encapsulation in 10 FG. Tissues then were cultured for 1 week in static or the non-static systems. Ovarian tissue quality was evaluated on day 0 and day 7 in terms of follicle viability (neutral red staining), follicle density and developmental stages (histology), and Akt phosphorylation (pAkt) (Western blot analysis).
3. Ovarian tissue retrieval
Ovaries from 31 female cats (age range, 1–3 years) without any pathological conditions (e.g., ovarian cysts, para-ovarian cysts, cystic endometrium hyperplasia, or pyometra) were included in the study. Tissues were collected from the routine ovariohysterectomy at the Veterinary Public Health Division of Bangkok Metropolitan Administration, Thailand. The collection and use of tissues were approved by Institutional Animal Care and Use Committee, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand. Ovaries were kept in isotonic saline solution supplemented with 1 IU/mL penicillin-streptomycin and transported to the laboratory at room temperature within 3 hours.
Cortical slices (~ 0.2×0.2×0.1 cm; width×length×thickness) were dissected from the ovaries using tissue scissors, scalpel blade and stored (38.5°C) in the maintenance medium (Eagle’s Minimum Essential Medium [MEM] containing 2 mM L-glutamine, 10 IU penicillin G sodium, 10 µg/mL streptomycin sulfate, 1 mg/mL bovine serum albumin, and 25 mM [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; HEPES]) for less than 1 hour prior to encapsulation and cryopreservation procedures.
4. Ovarian tissue encapsulation
Ovarian tissue encapsulation by fibrin clot construction was performed according to previous studies [4,14]. Briefly, ovarian cortical pieces were directly placed in fibrin gel drops (fibrinogen/thrombin cocktail) composed of 5 mg/mL fibrinogen and 50 IU/mL thrombin (5 FG group) or 10 mg/mL fibrinogen and 50 IU/mL thrombin (10 FG group) supplemented with 0.1 ng/mL VEGF. To create the fibrinogen/thrombin cocktail, the fibrinogen was diluted in 25 mM HEPES-0.9% saline solution, whereas the thrombin was prepared in 40 mmol/L CaCl2 solution. Ovarian tissues were placed in the hydrogel drops and then incubated at 37°C for 30 minutes to allow gel polymerization.
5. Ovarian tissue cryopreservation
Each encapsulated ovarian cortical piece was randomly allocated to slow freezing or vitrification procedures, as previously reported [15,16]. For slow freezing, ovarian tissues with each designed fibrin gel concentration, including the fresh control, were equilibrated in the freezing medium (MEM medium supplemented with 0.1 ng/mL VEGF, 1.5 M dimethyl sulfoxide [DMSO; Fluka Chemie, Buchs, Spain] and 0.1 M sucrose) at 4°C for 15 minutes, transferred to pre-cooled 2-mL cryovials (cryogenic vials; Corning Inc., Corning, NY, USA), and stored at 4°C for another 15 minutes. Cryovials containing equilibrated ovarian cortical slices were then kept in a freezing container (Mr. Frosty Freezing Container; Thermo Fisher Scientific, Waltham, MA, USA) at –80°C for 24 hours (cooling rate: –1°C/min) and manually dropped into liquid nitrogen (–196°C). For thawing, cryovial tubes were transferred to a water bath at 37°C for 5 minutes. The thawed ovarian tissues were then placed in freshly prepared media; first in MEM with 0.75 M DMSO and 0.25 M sucrose (room temperature for 10 minutes), then in MEM with 0.25 M sucrose (room temperature for 10 minutes), and finally in MEM alone as the maintenance medium.
For vitrification, control and encapsulated ovarian tissues were sequentially placed in (1) the equilibration medium (MEM with 20% fetal bovine serum [FBS] [v/v], 0.1 ng/mL VEGF, 0.96 M DMSO, and 1.21 M ethylene glycol [EG]; room temperature for 10 minutes); (2) the vitrification medium (MEM with 20% FBS, 0.1 ng/mL VEGF, 1.92 M DMSO, 2.42 M EG, and 0.5 M sucrose; 4°C for 30 minutes), and then transferred to pre-cooled cryovials and immediately plunged into liquid nitrogen. For warming, cryovials containing vitrified ovarian tissues were held in air at room temperature for 30 seconds. Ovarian tissues were placed in the warming medium (MEM with 1.0 M sucrose and 20% FBS; 37°C for 10 minutes), and in MEM alone as the maintenance medium.
6. Ovarian tissue culture
Fresh and cryopreserved cortical slices were randomly allocated to either the static [17] or the non-static culture system. For the static culture system, ovarian tissues were cultured on top of 1.5% (w/v) agarose gel submerged in 500 μL of culture medium (MEM supplemented with 2 mM glutamine plus 10 IU/mL penicillin G sodium, 10 µg/mL streptomycin sulfate, 0.05 mM ascorbic acid, 0.5 μg/mL insulin, 0.4 μg/mL transferrin, 0.5 ng/mL selenium, 10 ng/mL follicle-stimulating hormone, and 0.1% [w/v] polyvinyl alcohol) at 38.5°C in humidified 5% CO2 in air for up to 1 week. The culture medium was fully replaced every other day throughout the culture period. For the non-static culture system, a 12-well culture plate was modified by adding a 0.16-cm inlet tube connecting with the syringe pump (Chemyx, Stafford, TX, USA) and a 0.16-cm outlet port (Figure 2). A total of 20 mL of culture medium was loaded into the pump, which provided a constant flow rate (0.000173 mL/min) to the modified culture plate for 1 week. The constant flow rate was calculated from the volume of the culture medium in the static culture system that was refreshed every second day divided by 2,880 (48 hours multiplied by 60 minutes); the constant flow rate of 500 μL/2,880 min was therefore equivalent to 0.000173 mL/min.

Modified non-static culture system using 12-well culture plate connected with a 0.16-cm inlet tube-syringe pump (constant flow rate) and a 0.16-cm outlet tube.
7. Follicle viability and morphology assessments
The viability of follicles enclosed in the ovarian cortex was evaluated by neutral red staining [16]. Briefly, cortical slices were placed in the culture medium supplemented with 50 μg/mL neutral red and incubated in dark at 37°C for 1 hour. Follicular viability was assessed under the light microscope (Nikon Eclipse E100; Nikon, Tokyo, Japan) at ×100 magnification. All follicles within each cortical piece were counted and classified as viable (red color) or non-viable (non-staining) to calculate the percentage of viable follicles.
To estimate the developmental stage (percentage and ratio), the structural morphology and the density of follicles, ovarian tissues were fixed in 4% paraformaldehyde at room temperature and dehydrated in graded series of ethanol solutions prior to tissue sectioning [18]. Serial sections of each cortical piece (5 µm thickness each) were stained with hematoxylin and eosin. Two sections at 10-µm intervals of each cortical piece were evaluated under light microscopy at ×100 and ×400 magnification. The developmental stage was classified according to one of three categories: (1) primordial follicle (one layer of flattened granulosa cells circumscribing the oocyte); (2) transitional follicle (one layer of flattened and cuboidal granulosa cells surrounding the oocyte), or (3) primary follicle (one layer of cuboidal granulosa cells surrounding the oocyte). Follicles with intact oocytes and granulosa cells were categorized as having normal morphology in contrast with their abnormal counterparts with fragmented, pyknotic nuclei or disruption of granulosa cells [17]. Furthermore, the structural morphology of isolated follicles from the cortical tissue (left from neutral red staining) was assessed using 2 μM Hoechst 33342 staining (Invitrogen, Thermo Fisher Scientific) under a fluorescence microscope at ×200 magnification (Nikon Eclipse TI fluorescence microscope). To measure the follicle density, the number of the intact follicles in 10 random microscopic reticule areas (0.0625 mm2 per scale) were counted at ×100 magnification. The relative follicle density per 1 mm2 was calculated from the microscopic reticule areas and recorded.
8. Evaluations of Akt, Tie1, and VEGF-R phosphorylation
The ovarian tissues were minced using a mortar and were incubated with a protein lysis buffer (CelLytic MT) supplemented with a protease inhibitor for 30 minutes on a shaker at room temperature (22°C). After centrifugation at 12,000 × g at 4°C for 10 minutes, the supernatant was assessed for total protein concentration using NanoDrop One (Thermo Fisher Scientific) and adjusted to 15 µg/µL in phosphate-buffered saline before storage at –80°C. The Western blot analysis was performed as previously reported [19]. Briefly, post-thawed protein samples were mixed with SDS sample loading buffer (4× Laemmli sample buffer and β-mercaptoethanol; Bio-Rad, Hercules, CA, USA) and processed for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (4%–15% Mini-protein TGX Precast Gels; Bio-Rad) for 1.5 hours. The precision Plus Protein Dual Color Standards (Bio-Rad) loaded into the first lane were used as the molecular weight standard. The complete SDS-PAGE run was transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore, Billerica, MA, USA) and blocked (non-specific binding protein) with 2.5% skim milk (Bio-Rad) diluted in washing buffer (Tris-buffered saline plus 0.1% [v/v] Tween) for 1 hour at room temperature (22°C). The transferred membrane was then incubated on the shaker in the dark at 4°C overnight with 1 of the following primary antibodies: (1) rabbit anti-β-actin (1:1,000); (2) rabbit anti-phospho-Akt (1:1,000; Ser473 pAkt); (3) rabbit anti-Tie1 (1:100; Tie1 D2K2T) or IV) rabbit anti-phospho-VEGF-R-2 (1:1,000; Tyr1175). The sample incubated with the normal rabbit IgG (1:1,000) served as the negative control. After overnight incubation with the primary antibody, the blot membrane was incubated with the secondary antibody (anti-rabbit IgG antibody conjugated with horseradish peroxidase, 1:1,000) for 2 hours at room temperature (22°C). Immunoreactivity was detected by the colorimetric method (Opti 4CNTM Substrate Kit, Bio-Rad). The intensity of each protein band and background was measured using the Axygen Gel Documentation System (Corning, Corning, NY, USA). The relative quantification was normalized by dividing the intensity of each primary antibody (intensity of each protein band minus background) by that of the β-actin band.
9. Statistical analysis
Data analyses were performed using SAS ver. 9.2 (SAS Institute, Cary, NC, USA). The normal distribution of residuals was tested using the univariate procedure with the “normal” option. The dependent variables included follicle viability, normal morphology, follicle developmental stage, follicle density, and relative quantification of protein bands. When the data were normally distributed, differences in dependent variables among the experimental groups were compared using one-way analysis of variance followed by the Tukey-Kramer test. Non-normally distributed data were analyzed using the Wilcoxon signed rank test. Relative quantification of protein bands was evaluated using a mixed model (the “mixed” procedure). All results are presented as mean±standard deviation (SD). The level of significance was set at p≤0.05.
Results
1. Experiment I: influence of ovarian tissue encapsulation on cryopreservation outcomes
More than 80% of follicles were viable in the freshly-excised ovarian cortices. Although the values were similar (p>0.05) during equilibration, viability decreased (p≤0.05) during cryopreservation (Table 1). Follicle viability was influenced by the cryopreservation procedure, and vitrification without tissue encapsulation yielded the lowest percentages (p≤0.05) (Table 1). Although not significantly different from 5 FG encapsulation, the samples processed with 10 FG encapsulation before slow freezing or vitrification had better percentages of viable follicles (p≤0.05) than their non-encapsulated counterparts. However, slow freezing led to the highest number of viable follicles (p≤0.05) (Table 1).
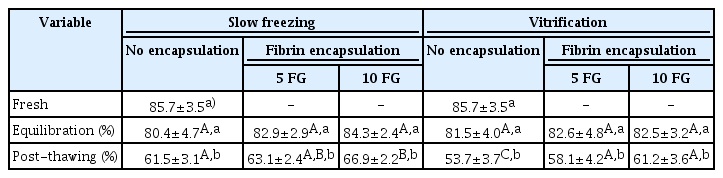
Percentages of viable follicles in ovarian tissues that were encapsulated or not encapsulated before equilibration and cryopreservation (slow freezing vs. vitrification)
In accordance with the viability observations, the histology revealed a decrease in the percentage of structurally normal follicles after cryopreservation (p≤0.05) (Table 2). The proportion of normal follicle morphology in the cryopreserved non-encapsulated tissues (control) was lower (p≤0.05) than in the encapsulated samples (Table 2, Figure 3). Although not significantly different from 5 FG encapsulation, the samples processed with 10 FG encapsulation and slow freezing presented the highest proportions of normal follicles (Table 2). When the isolated follicles were stained using the fluorescence dye, follicles with abnormal structural morphology were present in the fresh samples and all freezing conditions (Table 2). In the post-thawing evaluation, the percentage of structurally normal follicles in the control was lower than in the encapsulated samples (slow freezing group) (p≤0.05). As with the previous evaluation criteria, cryopreservation procedures negatively affected follicle density (p≤0.05) (Table 3). However, encapsulation could not mitigate the loss (Table 3). The best follicle density after thawing tended to be from 10 FG encapsulation and slow freezing (Table 3).
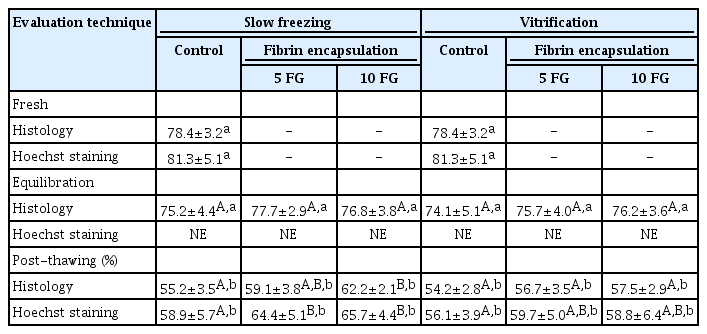
Percentages of morphologically normal follicles in ovarian tissues that were encapsulated or not encapsulated before equilibration and cryopreservation (slow freezing vs. vitrification)

Normal follicle morphology (one layer of flattened granulosa cells circumscribing the oocyte; arrow) retrieved from encapsulated ovarian cortex (A) compared to abnormal follicle morphology (disruption of granulosa cells; arrow) from the non-encapsulated group (B). bar=50 μM, H&E.
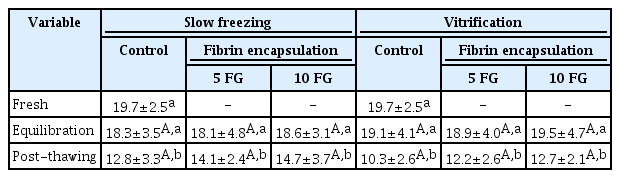
Follicle density (number of normal follicle morphology per 1 mm3) in ovarian tissues that were encapsulated or not encapsulated before equilibration and cryopreservation (slow freezing vs. vitrification)
The Western blot analysis revealed appreciable levels of Tie relative band intensity in the fresh controls (mean±SD, 0.32±0.09) and in all treatment groups (range, 0.24–0.32). In the 5 FG group, the Tie1 relative band intensity was not significantly different (p>0.05) between fresh controls and all treatment groups (range, 0.24–0.27). In the 10 FG group, the Tie relative band intensity ranged from 0.25 to 0.31 and also was not significantly different between the controls and the treatment groups (Figure 4A). For the VEGF-R phosphorylation relative band intensity, the values in the fresh and treatment groups ranged from 0.35 to 0.58 (fresh sample: mean±SD, 0.51±0.1). In the 5 FG group, the relative band intensities were not significantly different (p>0.05) between the fresh controls and all treatment groups (range, 0.37–0.41). In the 10 FG group, the VEGF-R phosphorylation relative band intensity in the encapsulated cortical tissues that were cryopreserved by slow freezing (mean±SD, 0.58±0.06) was significantly higher (p≤0.05) than in the non-capsulated samples (mean±SD, 0.42±0.09) and the ovarian tissues cryopreserved by vitrification (mean±SD, 0.35±0.08) (Figure 4B). The western blot bands revealed Tie1 and VEGF-R phosphorylation from selected animal were demonstrated in Figure 4C.
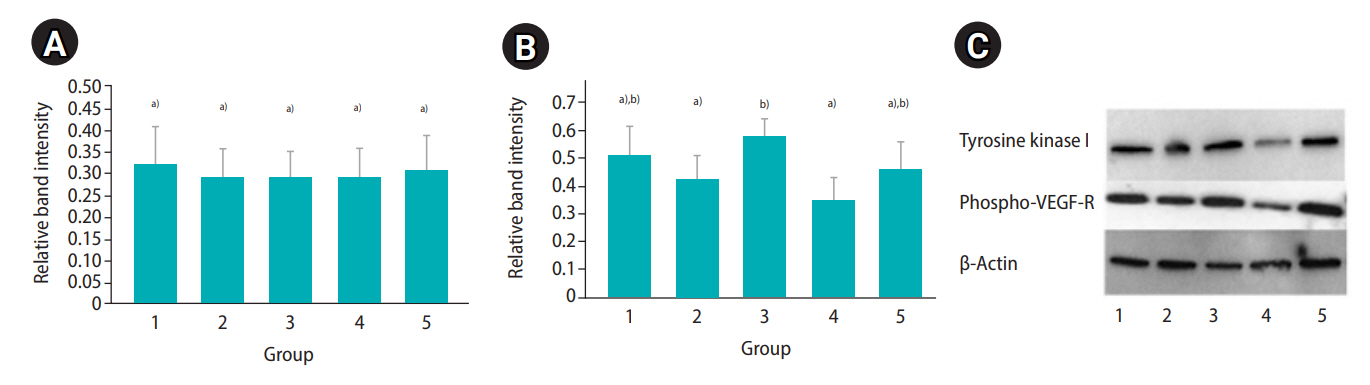
Relative band intensity of tyrosine kinase I (A) and vascular endothelial growth factor receptor (VEGF-R; B) phosphorylation in fresh (control), slow frozen, or vitrified cortical samples after encapsulation (with or without 10 mg/mL fibrinogen, 50 IU/mL thrombin, and 0.1 ng/mL VEGF; 10 FG) (mean±standard deviation; n=21 animals). Group 1, fresh ovarian tissue; group 2, ovarian tissue cryopreserved by slow freezing (without encapsulation); group 3, ovarian tissue cryopreserved by slow freezing (with encapsulation); group 4, ovarian tissue cryopreserved by vitrification (without encapsulation); group 5, ovarian tissue cryopreserved by vitrification (with encapsulation). (C) Western blot bands represent tyrosine kinase I and VEGF-R phosphorylation, signaling protein expression from a selected animal. a),b)Different letters indicate statistically significant differences between groups (p≤0.05).
Overall, the outcomes were better with 10 FG encapsulation than with 5 FG encapsulation. Therefore, this treatment was selected for further exploration in experiment II.
2. Experiment II: influence of static and non-static culture systems following encapsulation and cryopreservation on ovarian tissue integrity and survival
A decrease in follicle viability was detected after both slow freezing and vitrification (p≤0.05) while more than 50% of follicles remained viable (Table 4). The culture systems or the freezing protocols did not markedly influence follicle viability and density (Table 4). Importantly, the histological analysis demonstrated that encapsulation and culture systems were able to maintain proportions of follicular stages that were similar to those of the fresh controls (Figure 5). The ratio to primordial follicles (on day 0 [D0]) to transitional stage follicles (on day 7 [D7]) slightly varied depending on the condition (1.0:0.91 for the slow freezing and static culture system, 1.0:0.97 for the slow freezing and non-static culture system, 1.0:1.48 for the vitrification and static culture system, and 1.0:1.07 for the vitrification and non-static culture system). However, the average ratio of primordial follicles (D0) to primary follicles (D7) was 1.0:0.40 across all experimental groups (1.0:0.40 for the slow freezing and static culture system, 1.0:0.43 for the slow freezing and non-static culture system, 1:0.36 for the vitrification and static culture system, and 1.0:0.40 for the vitrification and non-static culture system). The phosphorylation of Akt was observed in all fresh control and treatment tissue samples (Figure 2). The relative quantification of pAkt band intensity did not significantly differ among groups (range, 0.19–0.25; p≤0.05) (Figure 6).
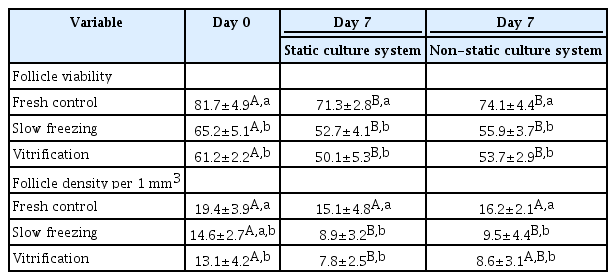
Percentages of viable follicles and follicle density (number of normal follicle morphology per 1 mm3) in ovarian tissues that were encapsulated (10 FG), cryopreserved (slow freezing vs vitrification), and cultured (static and non-static culture systems) for 7 days
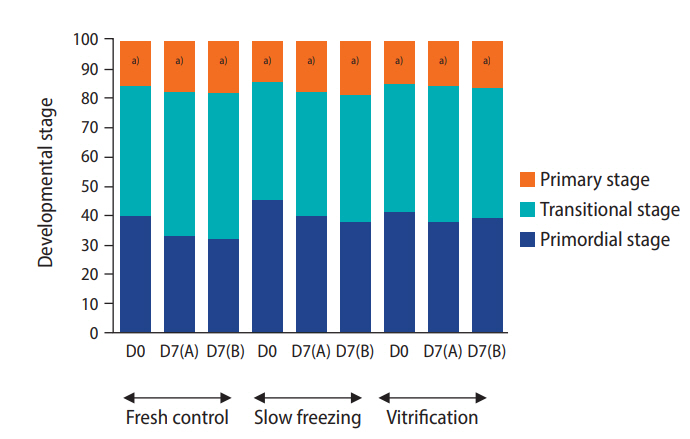
Mean percentage of follicles at each developmental stage in ovarian tissues that were freshly-excised (fresh control) or encapsulated (10 mg/mL fibrinogen, 50 IU/mL thrombin, and 0.1 ng/mL vascular endothelial growth factor; 10 FG), cryopreserved by slow freezing or vitrification procedures, and cultured for 7 days. D0, day 0; D7(A), static culture system for 7 days; D7(B), non-static culture system for 7 days (n=10 animals). a)Different letters indicate statistically significant differences in primary follicle development between groups (p≤0.05).
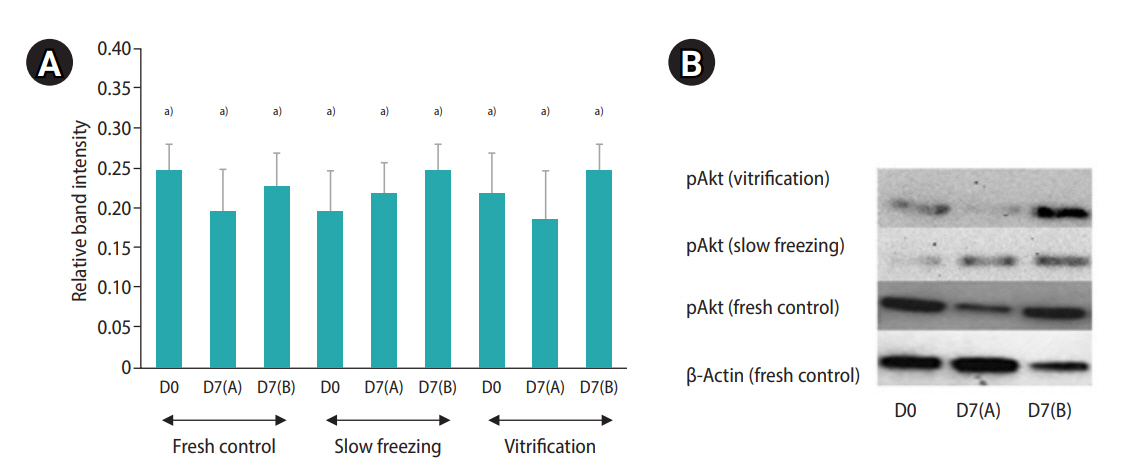
(A) Relative band intensity of protein kinase B phosphorylation (pAkt) expression in fresh (control), slow freezing and vitrification cortical samples (with or without 10 mg/mL fibrinogen, 50 IU/mL thrombin, 0.1 ng/mL vascular endothelial growth factor; 10 FG) and cultured in various conditions; D0, day 0; D7(A), static culture system for 7 days; D7(B), non-static culture system for 7 days (mean±standard deviation; n=10 animals).a)Different letters indicate statistically significant differences between each group (p≤0.05). (B) Western blot bands represent pAkt signaling protein (60 kDa) expression from a selected animal.
Discussion
Using domestic cats as a model for human fertility preservation, the combination of 10 FG encapsulation and slow freezing appeared to be the best combination to preserve follicle viability, normal morphology, and VEGF protein phosphorylation. Fibrin encapsulation also promoted post-thawing survival and the maintenance of different follicular stages during 7 days of culture (regardless of the culture system).
In 2017, a meta-analysis of 14 studies related to human ovarian tissue cryopreservation protocols reported non-significant differences between slow freezing and vitrification based on the proportion of intact primordial follicles [5]. However, ovarian tissue cryopreservation by slow freezing remains the most frequent technique in humans compared to vitrification methods [20]. Furthermore, slow freezing of ovarian tissue combined with transplantation has enabled more live births than vitrification [20]. Following human ovarian tissue cryopreservation and xenotransplantation, recent research data also revealed better outcomes with slow freezing than vitrification in terms of primordial follicle survival, primordial follicle counts, follicle growth, proliferation and angiogenesis markers (Ki-67 and CD31) [21]. In addition, the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and transmission electron microscopy showed higher levels of DNA damage and follicle deformation in the vitrification group [21]. Our findings are consistent with this previous study [21] since the percentages of the post-thawed follicle viability were lowest in the non-encapsulated vitrified cortical samples. Detrimental factors, such as an improper cooling rate and/or adverse effects of cryoprotective agents (CPA), have been shown to lead to follicle deformation, oocyte shrinkage, or granulosa cell disruption during ovarian tissue cryopreservation [22]. In the present study, the ovarian tissues were processed at a 2-fold higher concentration of CPA (15% DMSO) during vitrification protocol than during slow freezing (7.5% DMSO). The most up-to-date evidence as of 2019 markedly revealed the toxicity of DMSO when the DMSO concentration gradually increased (10% to 50%) during ovarian tissue cryopreservation by vitrification [23]. The resultant cellular membrane disintegration, chromatin thickening, and cell apoptosis induction likely led to follicle death [23].
Due to their unique properties, soft biodegradable materials or hydrogels have recently been explored and used in cell and tissue research [24]. In reproductive medicine, studies focused on isolated follicles encapsulated under the hydrogel micro-environment have been well documented [25,26]. However, reports on ovarian tissue enclosed in those novel materials are still limited. Our results notably demonstrated that fibrin encapsulation promoted favorable follicle morphology and survival after slow-freezing and thawing. Due to its typical 3D network structure and density, a porous hydrogel is typically adjustable, which allows it to have a proper affinity in the aqueous environment. It also mitigates changes of the osmotic stress caused by CPA exposure [6]. In addition, hydrogels can bind to the surface of very small ice crystals and prevent their detrimental expansion [6,27]. Therefore, hydrogel-based encapsulation has been considered an effective approach to preserve cells during cryopreservation [24]. This was confirmed in mouse immature testicular tissues encapsulated in a fibrin hydrogel [28]. Fibrin gel could serve as an integral component of the cryopreservation medium, which effectively contributed to the preservation of the average density of germinative cells and their metabolic activity (total lactate dehydrogenase activity) [28]. In addition to vitrification, fibrin encapsulation supported post-thawed follicle viability compared to non-encapsulated samples. Nonetheless, the observed results were lower than in the slow freezing group. In addition to the results related to the effects of CPA concentration, as discussed above, a previous study in c3h10t1/2 mesenchymal stem cells demonstrated that alginate encapsulation could significantly augment post-thawed cell viability when a cooling rate of more than 50,000°C/min was applied during the vitrification procedure by using a funnel-shaped quartz microcapillary as a freezing device [29]. Furthermore, the aqueous volume of the confined space around the cell should be less than 0.1 μL, which leads to the effective regulation of ice nucleation and water transport properties during vitrification [30]. Due to the complexity of the ovarian tissue compartment, the obtained results might be different from single-cell investigations. In addition, neither the cooling rate nor aqueous volume was measured in the present study. These might be among the principal factors that could detrimentally affect post-thawed follicle quality in ovarian tissue vitrification, as observed in our study.
Regarding protein phosphorylation results, the highest VEGF-R phosphorylation was observed in samples that underwent 10 FG encapsulation and slow freezing, while Tie1 phosphorylation did not differ among treatment groups. Along with their influential effects on cell integrity preservation, another crucial feature of fibrin hydrogels at gradually increasing concentrations (i.e. 12.5 or 25 mg/mL) is their support of angiogenic activity (i.e., VEGF-R) [31,32]. This is related to the presence of arginine-glycine-aspartate peptides, which can effectively promote crosslinking with various types of exogenous angiogenic or growth factors [31,32]. Furthermore, the fibrin scaffold could potentially mimic physiological angiogenesis, immobilize VEGF in the matrix, and prolong and activate its release, which might lead to VEGF-R activation [33,34]. Supporting data were found in a mouse ovarian tissue transplantation study, in which the post-thawed cortical piece encapsulated with the fibrin hydrogel modified with heparin-binding peptide (HBP) and VEGF had the greatest number of viable primordial follicle count per tissue section (follicle density) compared to non-encapsulated or fibrin-HBP samples [34].
For Tie1, this receptor is structurally related to tyrosine kinase receptors, which are selectively expressed on vascular endothelial cells, promote vascular integrity, and are considered to be an endothelial cell activation marker [8]. However, data about VEGF-Tie1 activity related to ovarian function remain limited. A study of human umbilical vein endothelial cells noted that VEGF could stimulate the formation of the Tie1 endodomain during a 30-minute VEGF treatment, whereas it did not increase in response to 24-hour VEGF stimulation [35]. In addition, tumor necrosis factor-alpha (TNFα), a relevant marker of apoptotic cell death that increases during mammalian cell cryopreservation, could stimulate Tie1 protein cleavage and decrease the cellular Tie1 activity over 24 hours [35]. In our study, the imbalance between VEFG and TNFα activities might primarily explain the absence of a difference in Tie1 protein phosphorylation among all frozen groups due to the short exposure of VEGF to the ovarian samples prior to the cryopreservation process. To obtain stronger evidence, the dynamic regulation of TNFα during encapsulated ovarian tissue cryopreservation should be further investigated.
In recent research, dynamic microenvironment circulation in non-static culture systems using precise media flow rates (microfluidics) was employed and offered an innovative technology for single or multiple cells/tissues in long-term culture [9]. In our study, the best follicle quality was observed in the ovarian cortices cultured in the non-static system for 1 week. However, the data did not differ statistically from the control group. Previous studies reported the efficacy of dynamic culture media flow for maintaining gamete cell development and hormone production [9,10]. For example, neonatal mouse testicular tissues cultured in a microfluidic platform presented the distribution of green fluorescence protein (GFP) throughout the tissues, whereas GFP was absent in the agarose gel block tissue cultured under the static system [36]. Additionally, in observations after a 39-day dynamic culture system, round and elongated spermatids were perceived with GFP cap-like the arched structures, providing evidence of spermatid development [36]. In female reproductive biology research, a study in 2020 performed follicle activation in cat ovarian tissue culture under a microfluidic chamber of soft-lithography polydimethylsiloxane (PDMS) [37]. The results showed that viable and normal structural follicles could be maintained in the microfluidic device after 4 days, unlike samples on an agarose gel block [37]. Our contradictory findings may have resulted from differences in the tissue culture system (with or without an agarose gel block), culture media flow rate (0.002 mL/min [37] vs. 0.00017 mL/min in our study), or the type of device fabrication (3D printing-PDMS or a modified conventional cell culture plate). Although gonadal tissue culture on agarose gel blocks has been attempted [17,38], recent evidence showed a diminished distribution of GFP protein markers related to spermatid development during long-term mouse testicular tissue culture using agarose gel [36]. With more than a 10-times lower flow rate [37], the oxygen and nutrients diffused through the agarose gel and the hydrogel encapsulation might have been insufficient [35]. Consequently, precise microfluidic platform construction also requires expert-level device fabrication [10,35]. In the present study, the basic design of the non-static culture platform was preliminary and manually adjusted based on the conventional cell culture plate with a flexible size of inlet/outlet ports. Hence, less precise tissue culture compartments were conceivably made compared to the microfluidic device fabricated by 3D printing technology. Finally, the present findings did not reveal a difference in pAkt relative band intensity among groups, supporting the non-significant results for follicle stage and development. Although pAkt can trigger the phosphorylation of the transcription factor forkhead box O3, which is an important cell proliferation and differentiation regulator, the phosphorylation of Akt can be diminished by various factors, such as epigenetic regulation [39]. Recent data in 2018 demonstrated that the overexpression of non-coding RNA (microRNA), such as miRNA-195, activated interleukin and TNFα, while suppressing protein expression (VEGF, PI3K, and pAkt) [40]. Additionally, alterations of microRNA have been observed during cell culture and cryopreservation procedures [41]. Further expanded studies on genetic/epigenetic regulation and protein expression should be considered to augment the missing data in the present study.
In conclusion, our findings demonstrated the benefits of novel technologies focused on ovarian tissue encapsulation, cryopreservation, and in vitro culture by a non-static system using a cat model. Our study examined follicle viability, the percentage of normal morphology, and protein phosphorylation during tissue encapsulation and freezing procedures. These findings could potentially be translated to fertility preservation in cancer patients along with other potential modernized steps of ovarian tissue cryopreservation and culture strategies. Additionally, the present data may contribute to the achievement of IVA, leading to IVG and retransplantation of follicles without malignant cancer cell contamination.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Author contributions
Conceptualization: PT (Paweena Thuwanut), AP, WS (Weerayut Srituravanich). Data curation: PT (Paweena Thuwanut), WS (Weerayut Srituravanich). Formal analysis & Funding acquisition: PT (Paweena Thuwanut). Methodology: PT (Paweena Thuwanut), WS (Weerayut Srituravanich). Project administration: PT (Paweena Thuwanut), KP. Visualization: WS (Wisan Sereepapong), CT, CS, PT (Punkavee Tuntiviriyapun). Writing–original draft: PT (Paweena Thuwanut). Writing–review & editing: PC, PS.
Acknowledgements
The authors were grateful to the veterinary surgery team for reproductive organ collection.