Autophagy in the uterine vessel microenvironment: Balancing vasoactive factors
Article information

Abstract
Autophagy, which has the literal meaning of self-eating, is a cellular catabolic process executed by arrays of conserved proteins in eukaryotes. Autophagy is dynamically ongoing at a basal level, presumably in all cells, and often carries out distinct functions depending on the cell type. Therefore, although a set of common genes and proteins is involved in this process, the outcome of autophagic activation or deficit requires scrutiny regarding how it affects cells in a specific pathophysiological context. The uterus is a complex organ that carries out multiple tasks under the influence of cyclic changes of ovarian steroid hormones. Several major populations of cells are present in the uterus, and the interactions among them drive complex physiological tasks. Mouse models with autophagic deficits in the uterus are very limited, but provide an initial glimpse at how autophagy plays a distinct role in different uterine tissues. Herein, we review recent research findings on the role of autophagy in the uterine mesenchyme in mouse models.
Introduction
The uterus is composed of multiple cell types that respond distinctively to hormones and growth factors [1]. The epithelial lining, the innermost layer facing the lumen, is where the embryo attaches for implantation. The underlying stroma contains a sparse population of fibroblasts that secrete extracellular matrix proteins to fill the space among blood vessels and lymphatic vessels. The stroma contains numerous capillaries and spiral arteries, as well as diverse types of leukocytes, in the deep regions. The outermost layer, the myometrium, consists of smooth muscle cells; the uterine length and diameter can be regulated by coordinated actions of longitudinal and circular layers of smooth muscle cells. The uterine capillaries are surrounded by a single endothelial layer, similar to other capillaries found throughout the body, but larger vessels found in deep stromal regions and the myometrium are also surrounded by vascular mural cells (i.e., smooth muscle cells and pericytes). Blood flow to the uterus and vascular permeability are thought to be regulated by ovarian steroid hormones and vasoactive factors [2].
As a dynamic organ that responds to cyclic changes of the reproductive cycle and pregnancy, the uterus experiences tissue remodeling occurring at regular intervals. The uterus also ages with waning levels of steroid hormones depending on the species. Multiple female-specific pathologic conditions involving the uterus exist that are related or unrelated to pregnancy [3]. Its collective complexity makes the uterus a difficult-to-study organ system in gene-deletion studies. In this review, I attempt to present the possible outcomes of autophagic deficit in uterine tissue-specific mouse models based on previous studies and unpublished observations.
Autophagic process in brief
Three types of autophagic processes exist; microautophagy, macroautophagy, and chaperone-mediated autophagy [4]. Macroautophagy, the main bulk degradation pathway involved in turning over macromolecules and organelles, will hereafter be referred to as autophagy. The autophagic process is governed by proteins encoded by autophagy-related (Atg) genes, along with other associated factors [5]. The process begins with the formation of a horseshoe-shaped stretch of membrane near a target within the cytoplasm. This membrane grows with the addition of lipids from various subcellular sources until the two ends meet to form a closed vesicle called an autophagosome [6]. Initially, the contents within the autophagosome and double membrane structure are visible at the ultrastructure level [7]. Next, the autophagosome fuses with other vesicular structures such as endosomes or lysosomes. As degradation of the intravesicular contents begins, the double membrane structure and targets are no longer clearly visible. Therefore, it is customary to examine the ultrastructure of cells when attempting to demonstrate autophagic activation. This is considered the most direct evidence of autophagy [8].
Atg proteins and others involved in the pathway are subjected to post-translational modifications such as phosphorylation, acetylation, lipidation, and ubiquitination [5]. The most widely used method of visualizing heightened autophagy is to show the ratio of LC3B-I and LC3B-II changes in cells or tissues of interest on western blots. Pro-LC3B is first processed by Atg4 to LC3B-I, which, in turn, is lipidated by the Atg7-Atg3 complex. An increased ratio of LC3B-II/LC3B-I generally indicates an increased autophagic rate. Autophagy has a clear endpoint: the degradation of intravesicular contents and recycling. Trapped protein targets are tagged with a marker of destruction called sequestosome-1 (SQSTM1/p62) protein. SQSTM1 is a polyubiquitin-binding protein that links protein targets to autophagic degradation [9]. When autophagic flux is high and works well, the amount of SQSTM1 is maintained at a low level. In contrast, any problem that occurs in the autophagic process, such as the deletion of an important upstream Atg (Atg5 or Atg7) or defective fusion between an autophagosome and endosome/lysosome, results in the accumulation of SQSTM1 in cells. In mammalian cells, an increased ratio of LC3B-II/LC3B-I, along with a decrease in SQSTM1 levels under certain conditions, is considered to indicate increased autophagic flux [8].
Immunofluorescence staining of LC3B or SQSTM1 is also widely used, but interpreting the data requires caution. As autophagic vacuoles at different maturation stages form small vesicular structures within cells, the presence of LC3B-II on the membranes of autophagic vacuoles or SQSTM1 within them should exhibit puncta-like patterns in the cytoplasm, rather than a diffuse pattern. Extensive guidelines for the use and interpretation of autophagy assays are available [8].
Searching for a suitable in vivo model system
Various autophagy-deficient mouse models are available for in vivo studies [10,11]. Systemic deletion of Atg genes in core conjugation machinery generally leads to neonatal lethality, precluding the possibility of studying gene function in adult tissues and organs [11]. Tissue-specific knockout of floxed Atg7 using various Cre models has been widely used to study the physiological roles of autophagy [10,12].
Atg7, an E1 ubiquitin-activating enzyme, activates LC3B and Atg12 during autophagosomal membrane elongation. As one of the major components of the Atg conjugation system, the deletion of Atg7 shuts down autophagy during the initial stages of autophagosome formation, leading to neonatal lethality in knockout mice [12]. Atg5 is also a crucial component of the conjugation system, and its systemic deletion leads to neonatal lethality [13]. The Atg5 knockout mouse model was later further exploited in a study where the rescue of neonatal lethality was attempted by re-expressing transgenic Atg5 in neurons [14]. The neuron-specific re-expression of Atg5 in the Atg5 knockout background averted the neonatal death of the knockout mice, while still retaining an autophagic deficit in the rest of the body. In this model, a diverse array of organ abnormalities was observed. One remarkable phenotype was hypogonadism in both male and female transgenic mice [14]. The pituitary gland and gonads all showed high levels of SQSTM1 accumulation in these transgenic mice, suggesting that these organs heavily depend on autophagy for normal functions. The uteri of these Atg5 knockout mice were severely hypoplastic, which may be due to hypogonadism [14]. This study presented clear evidence that the reproductive hormonal axis requires normal autophagic flux to support fertility. Therefore, to establish how autophagy is involved in normal uterine functions, a uterine-specific knockout model was in demand.
Several cell type-specific expression models of Cre recombinase gene are available to study gene function in the uterus. Anti-Mullerian hormone type 2 receptor (Amhr2)-Cre is a knock-in model that expresses Cre in the uterine mesenchyme-derived cells only, except for a subset of cells on the mesometrial side [15]. The uterine epithelium and endothelial cells were excluded from gene deletion. Lactoferrin (Ltf) promoter-regulated Cre mice achieve floxed gene deletion only in the uterine epithelium [16]. Progesterone receptor (Pgr)-Cre is a knock-in model where Cre replaces 1 copy of Pgr [17]. This model shows the widest range of Cre expression in almost all cells present in the uterus, including endothelial cells [18] and various immune cells [19,20].
Our laboratory previously produced uterine tissue-specific deletion models of floxed Atg7 (Atg7f/f) with the aforementioned Cre models. Initial surveys searching for the site of high SQSTM1 levels in all three models revealed an interesting phenomenon. As we had already reported that hormone deprivation achieved by ovariectomy (OVX) turns on autophagy in the uterus [21], we surmised that OVX of these Cre mice would lead to SQSTM1 accumulation in cells where autophagy is active. Therefore, we compared the status of SQSTM1 accumulation in OVX mice in all three models. Amhr2-Cre/Atg7f/f mice showed high SQSTM1 accumulation in the uterine stroma and myometrium. As expected, some cells in the mesometrial side did not show SQSTM1 accumulation, as Atg7 was retained in that area (Figure 1A). This result indicate that autophagy is generally active in these mesenchymal cells. Pgr-Cre/Atg7f/f mice were expected to remove floxed Atg7 in all uterine cells including the epithelium. However, Pgr-Cre/Atg7f/f uteri showed high SQSTM1 accumulation in the stroma and myometrium, but limited accumulation in the epithelium (Figure 1B). This result suggests that the uterine epithelium does not depend on autophagy as much as the uterine mesenchyme. This notion was further demonstrated in the Ltf-Cre/Atg7f/f uteri, where SQSTM1 was almost undetectable in the Atg7-deleted uterine epithelium. A small population of luminal epithelial cells showed some puncta, as indicated by an arrow in Figure 1C. From these initial surveys, it became evident that the uterine mesenchyme heavily depends on autophagic clearance.
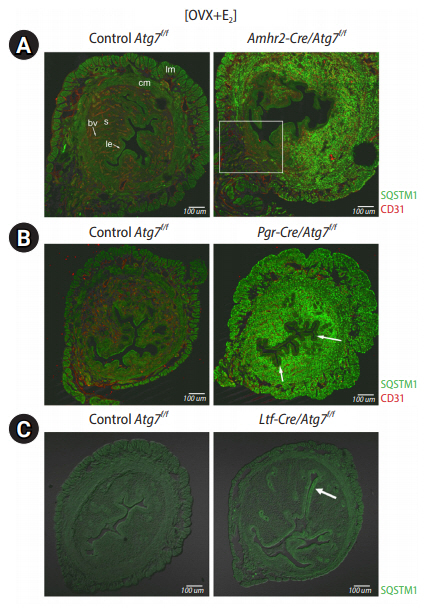
SQSTM1 accumulation in uterine-specific Atg7 deletion models after ovariectomy. Approximately 8- to 10-week-old mice were ovariectomized and rested for 2 weeks for hormone clearance. They received an injection of 17β-estradiol (E2) 24 hours before euthanasia. Uterine cross-sections were subjected to immunofluorescence staining with anti-SQSTM1 antibody. SQSTM1 signals are shown as green puncta in cells with autophagic deficit. Scale bar=100 μm. lm, longitudinal muscle; cm, circular muscle; bv, blood vessel; s, stroma; le, luminal epithelium. (A) Amhr2-Cre/Atg7f/f mice. Since the Amhr2 promoter is not active in some mesenchymal cells in the mesometrial side (white rectangle area), Atg7 is retained here. High SQSTM1 accumulation is shown in the stroma and myometrium, suggesting that autophagy is in demand in these cells. CD31, a marker of endothelial cells, was co-stained red. (B) Pgr-Cre/Atg7f/f mice. The Pgr promoter is active throughout the uterus. SQSTM1 accumulation is shown in the stroma and myometrium, but the epithelium does not show distinct accumulation. Some cells in the luminal epithelium show weak SQSTM1 accumulation (arrows). This suggests that the demand for autophagic turnover is much lower in the uterine epithelium than in the mesenchyme. (C) Ltf-Cre/Atg7f/f mice. The Ltf-Cre promoter is active in the uterine epithelium. Autophagic deficit is expected in the epithelium, but SQSTM1 accumulation is only weakly shown in some areas of the luminal epithelium (arrow).
Uterine vessel microenvironment demands autophagy
During reproductive cycles and pregnancy, progesterone and estrogen regulate the proliferation and differentiation of cells in a tissue-specific manner. Such cyclic changes produce diverse molecules with different functions, and dynamic turnover of these molecules is expected to occur to facilitate clearance. What are the pathophysiological outcomes of uterine cells in OVX mice when the autophagic process is entirely blocked? This question was addressed through the use of Amhr2-Cre/Atg7f/f mice, where the need for dynamic autophagic flux was demonstrated by high SQSTM1 accumulation in the uterine mesenchyme [22].
In various systems, the autophagic rate can increase beyond a basal rate when cells face certain changes within themselves or their surrounding environment, such as inflammation, viral infection, stress, hypoxia, or deprivation of nutrients [4,6,23]. Conditions affecting the autophagic rate vary greatly depending on the cell or tissue type. As a highly hormone-responsive organ, the uterus responds to OVX and increases autophagy in all major uterine cell types [21]. Systemic deprivation of steroid hormones also induces autophagy in other organs, such as the kidneys and prostate [24]. In Amhr2-Cre/Atg7f/f mice, accumulation of SQSTM1 in the uterine mesenchyme begins to be noticeable within 3 days after OVX (unpublished observation). In random-cycling Amhr2-Cre/Atg7f/f mice, SQSTM1 puncta begin to show in the mesenchyme of 4-week-old uteri at a much lower intensity than in OVX Amhr2-Cre/Atg7f/f mice [22]. Therefore, in the mouse uterus, hormone deprivation increases the autophagic rate, especially in the stroma and myometrium [21], as evidenced by high SQSTM1 accumulation in these tissues in OVX Amhr2-Cre/Atg7f/f mice. When steroid hormone levels are adequate during the reproductive cycle and pregnancy in intact mice, drastic increases in the autophagic rate probably do not occur.
The phenotype of the Amhr2-Cre/Atg7f/f model was further scrutinized to identify factors generally targeted by autophagy. Amhr2-Cre/Atg7f/f mice were found to be fertile, producing pups comparable to those of control mice. One outstanding characteristic of Amhr2-Cre/Atg7f/f uteri was that their stromal regions showed exaggerated edema [22]. Water imbibition in the uterus was significantly higher in the Amhr2-Cre/Atg7f/f uteri than in the control uteri, suggesting that the Amhr2-Cre/Atg7f/f uterine vessels are leakier and hyperpermeable. The uterine blood vessels were found to be more relaxed and leakier than those of control mice based on the expression levels of the endothelial junctional proteins [22]. Nitric oxide (NO) is a strong vasorelaxant produced by NO synthases (NOS). Among the three forms of NOS, NOS1 is present at notably increased levels in Amhr2-Cre/Atg7f/f uteri [22]. Based on these strong vascular phenotypes, it was surmised that autophagy is in strong demand for the maintenance of dynamic uterine vascularity.
Phenotype hints at the targets of uterine autophagy: vasoactive factors
The uterine mesenchymal cell populations can be roughly separated from the thick myometrium and uterine epithelium using the conventional method of uterine stromal cell (USC) preparations [25]. This method yields a heterogeneous cell population containing fibroblasts, smooth muscle cells, endothelial cells, immune cells, and vascular pericytes [25]. The vascular phenotype in Amhr2-Cre/Atg7f/f uteri prompted us to focus on the dysregulation of vasoactive factors in isolated USCs [22].
Vascularity is governed by various vasoactive factors. Vascular endothelial growth factor A (VEGFA) is an essential vasoactive factor in the uterus [18,26] that increases vascular permeability and dictates adult tissue angiogenesis in the mouse uterus under hormonal effects [2]. VEGFA was indeed identified as one of the proteins accumulated in Amhr2-Cre/Atg7f/f USCs. VEGFA sits at the top of the vasoactive regulatory pathway by regulating the function of other vascular factors. One of the deciphered pathways involves increasing the production of NO in the vessel microenvironment, thereby causing vessel relaxation and leakiness [27]. NO does this by nitrosylating the seminal endothelial junction stabilizing the protein β-catenin, leading to the disintegration of the endothelial barrier [28]. In Amhr2-Cre/Atg7f/f uteri, several observations have aligned to indicate the potential mechanism of uterine hyperpermeability: autophagic deficit leads to VEGFA overaccumulation, which, in turn, produces a greater amount of NO and, consequently, the disintegration of the endothelial barrier. The notion that NO is the mediator of hyperpermeability was demonstrated by the use of the NOS inhibitor, N-nitroarginine methyl ester (L-NAME). L-NAME administration to OVX Amhr2-Cre/Atg7f/f mice decreased stromal edema and restored the levels of β-catenin.
Physiological interventions: compensation of hyperpermeability in the absence of autophagy
While these results summarize what happens in the uterine mesenchyme in the absence of autophagy, other changes in Amhr2-Cre/Atg7f/f uteri have demonstrated remarkable compensatory changes to counterbalance the hyperpermeability phenotype [22]. The USC populations from Amhr2-Cre/Atg7f/f uteri consistently had a significantly greater number of cells than USCs from control mice. Among many cell populations, the melanoma cell adhesion molecule (MCAM/CD146)-positive population showed the most dramatic increase in USCs from Amhr2-Cre/Atg7f/f uteri compared to control USCs. MCAM is a broad mesenchymal marker [29,30] that is important for the maintenance of vascular permeability in the blood-brain barrier [31]. Therefore, an increased MCAM+ population in Amhr2-Cre/Atg7f/f uteri with leaky vessels could be an attempt to fortify vessels to alleviate the hyperpermeability phenotype.
Endothelin-1 (EDN1) is a potent vasoconstrictor expressed in many tissues, including the uterus [32]. In Amhr2-Cre/Atg7f/f USCs, the expression level of Edn1 mRNA is significantly decreased, whereas its protein levels are much higher than that in control USCs [22]. The heightened EDN1 levels would normally suggest a vasoconstriction phenotype, but vessels in Amhr2-Cre/Atg7f/f uteri remain relaxed. Therefore, EDN1 accumulation appears to be a compensatory mechanism to reduce overly relaxed vessels. However, in Amhr2-Cre/Atg7f/f uteri, overrepresented VEGF signaling appears to override EDN1 action.
Conclusion
The uterus, a dynamic organ with many functions, is constantly influenced by factors produced both outside of and within itself. The uterus undergoes cyclic changes in cell proliferation and differentiation, stromal edema, angiogenesis, and regeneration; however, it is resilient to all these changes. It is plausible that bulk degradation by autophagy is required for quality control in this versatile tissue. Compared to the uterine mesenchyme, the epithelium seems almost quiescent with respect to autophagic activation. Further investigation is warranted to reveal the mechanism of protein quality control in the uterine epithelium. In mice, gene-manipulated models generally provide the most direct evidence for gene function. In humans, where no such models are available, uterine and placental tissues obtained during various procedures have been used as the primary source for autophagy research. Review articles summarizing the potential role of endometrial or placental autophagy are recommended for further reading [33,34].
Acknowledgements
The author would like to thank the former and current members of the laboratory for their contributions.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.