A spontaneous pregnancy and live birth in a woman with primary infertility following the excision of an ovarian adrenal rest tumor: A rare case
Article information

Abstract
Adrenal rest tumors are a rare extra-adrenal complication of congenital adrenal hyperplasia (CAH) in women although they are more commonly found in the testes of male patients with CAH. An ovarian adrenal rest tumor (OART) may coexist with CAH or imitate its symptoms without CAH. In this case report, we present the case of a woman with OART without CAH, whose main complaint was infertility and who had a baby after successful surgical treatment.
Introduction
Ovarian adrenal rest tumors (OARTs) occur in a very small proportion (<0.1%) of women with congenital adrenal hyperplasia (CAH), and are extremely rare in women without CAH [1], although adrenal rest tumors are relatively common in the testes of men with CAH [2]. According to the literature, these tumors imitate CAH symptoms (e.g., hirsutism, clitoromegaly, oligo-amenorrhea, and, rarely, pelvic masses) [3]. Herein, we describe the first known case of OART in a woman without CAH presenting with primary infertility.
Case report
This is a case report, and the patient and her relatives were informed in detail and their written consents were obtained.
A 34-year-old nulliparous woman presented to our in vitro fertilization (IVF) center because of an inability to conceive for 2 years. While taking her medical history, the patient reported that her menstrual cycle had started at the age of 13, and was regular until the age of 19. Upon her first examination, the patient had been diagnosed with polycystic ovary syndrome due to her severe hirsutism and secondary amenorrhea (two of the three Rotterdam 2013 criteria). Therefore, she used oral contraceptive (OC) pills for 12 years. Her menstrual cycle improved while taking OC medication, and her hirsutism partially improved. She discontinued OC treatment after marriage because of a desire to have a child.
Two months after marriage, her irregular menses recurred. She presented to a local hospital with this complaint. Upon evaluation, her basal serum 17-hydroxyprogesterone (17-OHP) level was 20 ng/mL, which was higher than normal, leading the clinicians to consider the possibility of CAH. Therefore, dexamethasone therapy (0.5 mg/day) was started. Several ovulation inductions (with an aromatase inhibitor and timed coitus for six cycles) and an IVF cycle were performed with the goal of conception after the 6th month of her marriage. During IVF therapy, the gonadotropin (recombinant follicle-stimulating hormone [FSH]) dose used was 300 IU/day for 9 days and the endometrial thickness was 6.4 mm on trigger day. From the two collected oocytes, one mature oocyte was obtained and fertilized. The embryo was transferred on day 3, but she failed to achieve a pregnancy, even though her husband was healthy and his semen analysis was normal.
When she was transferred to us, we performed a physical examination of the patient. We assessed hirsutism using the Ferriman-Gallwey score, with a score of 22. There were also signs of virilism, including thickening of the vocal cords, alopecia, and clitoromegaly. The results of the hormone assay indicated that the patient’s 17-OHP level remained high (86 ng/mL), despite dexamethasone therapy. Her dehydroepiandrosterone sulfate, free testosterone, and total testosterone levels were 385.9 µg/dL, 5.5 pg/mL, and 416.2 ng/dL, respectively. Serum FSH, luteinizing hormone, thyroid-stimulating hormone, free T4, prolactin, and cortisol levels were normal. Transvaginal ultrasonography showed a normal uterus size, with a thin endometrium (4 mm) and diminished ovarian reserve. The right ovary measured 40×35×32 mm in size and had one antral follicle, and the left ovary measured 29×25×25 mm in size and had three antral follicles. A 34×30×29 mm solid mass was observed in the right ovary (Figure 1). Tumor markers (i.e., CA 125, CA 15.3, alpha-fetoprotein, and carcinoembryonic antigen) were found to be within normal limits. Since she had an ovarian mass and a high serum 17-OHP level, we suspected an adrenal rest tumor and ordered an abdominal and pelvic magnetic resonance imaging scan. It showed a 42×32-mm hyperintense right ovarian solid mass with a normal left ovary; however, the bilateral adrenal glands were of normal size and morphology (Figure 2). We planned laparoscopy for a definitive diagnosis of the right ovarian mass. Laparoscopic examination of the pelvic cavity showed a yellowish solid 4-cm mass on the right ovary. The mass was removed in an endoscopic bag without rupture (Figure 3), and sent for an immediate pathological frozen section, which revealed a benign tumor. Then it was sent for histological examination. Macroscopically, it was a yellow-colored tumor measuring nearly 4 cm, with a smooth and well-limited surface. Microscopically, almost all of the ovarian parenchyma was occupied by the tumor lesion, as shown by large cells with small nuclei without mitotic activity, nuclear atypia, and Reinke crystalloids. Hemorrhagic or necrotic areas were absent in the sample. Immunohistochemical analysis revealed diffuse staining for calretinin, inhibin, vimentin, and melan-A (Figure 4). The final histopathological findings confirmed the diagnosis of OART.
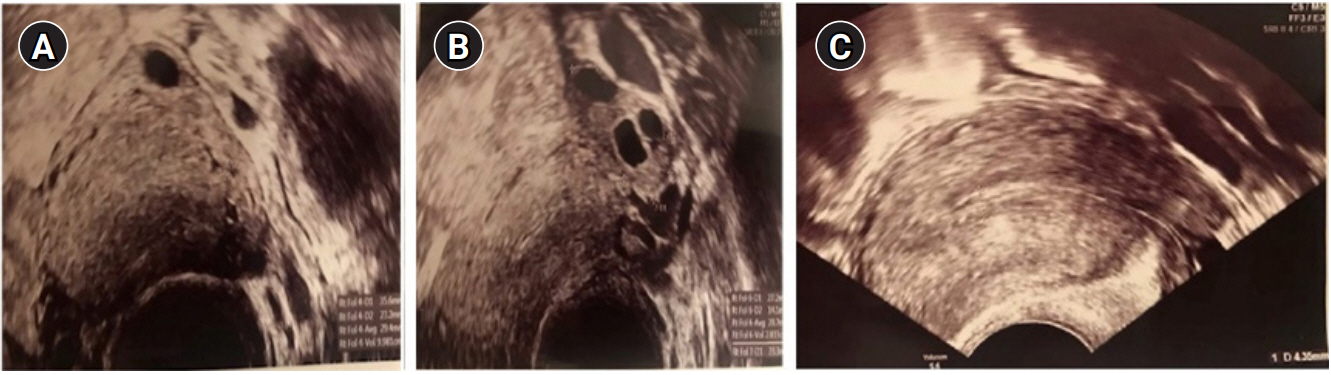
Ultrasonographic appearance of the right ovarian adrenal rest tumor demonstrating an echogenic border (A), the left ovary (B), and thin endometrium (C).
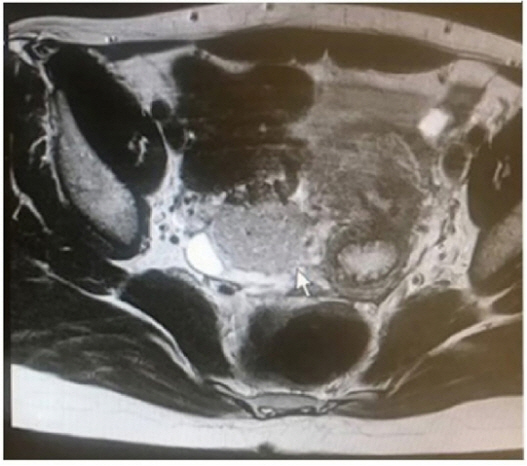
Magnetic resonance imaging showed a 42×32-mm hyperintense right ovarian solid mass in the pelvic cavity.
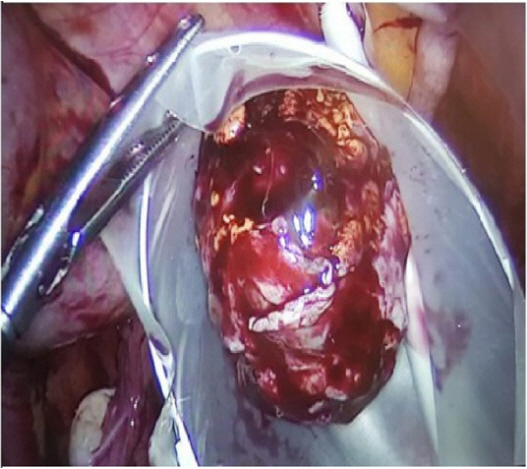
Laparoscopic removal of the yellowish solid mass in an endoscopic bag.
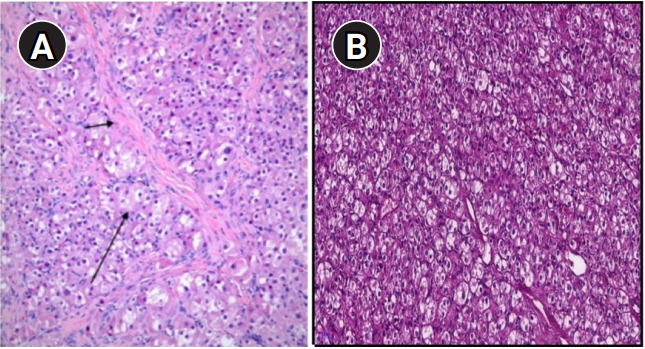
(A) Microscopic appearance of the tumor cells showed a uniform size, polygonal shape, abundant eosinophilic and vacuolated cytoplasm, and round nuclei with small nucleoli (long arrow). Acellular hyaline areas were observed among clusters of tumor cells (short arrow). (B) Mitotic activity was low. Crystalloids of Reinke were not identified (H&E, ×200).
One week after the operation, the patient’s serum 17-OHP level decreased dramatically (1.82 ng/mL). Therefore, we suspected CAH, and consulted with an endocrinologist to carry out an adrenocorticotropic hormone (ACTH) simulation (Synacthen) test for the definitive diagnosis of CAH. In the ACTH simulation test, the 17-OHP levels were 0.21 ng/mL, 0.54 ng/mL and 0.60 ng/mL at 0, 30, and 60 minutes, respectively. Based on these findings, the diagnosis of CAH was excluded. We also conducted a genetic test to investigate the possible presence of a CYP21A2 gene mutation, the test result was negative. We offered detailed genetic tests for the other enzyme deficiencies involved in CAH, but the patient did not agree to undergo the recommended tests.
Postoperatively, the patient’s menstrual cycles became regular and signs of hyperandrogenism disappeared. Furthermore, 4 months after the operation, she spontaneously became pregnant and then gave birth to a healthy infant at term.
Discussion
Adrenal rest tumors are benign, but may produce excessive 17-OHP and androgens. These tumors are commonly found in the testes of male patients, and their prevalence has been reported to be as high as 95% in men with CAH. In contrast, OARTs are less common in women with CAH (<0.1%) [1]. In a literature search, we found that Yilmaz-Agladioglu et al. [3] reported the first case of OART in a girl without CAH. They presented a 13-year-old female patient who was diagnosed with non-classical CAH at 6 years of age due to premature pubarche, and received hydrocortisone therapy for 6 years. The patient was further investigated because hormonal control was unsuccessful (high levels of 17-OHP and total testosterone) while under high-dose steroid therapy, and she was diagnosed with a steroid cell ovarian tumor, similar to our case. In our case, signs of hyperandrogenism, disordered menstruation, and infertility were the main complaints of the patient, whereas, in their case, the patient’s complaint was limited to signs of hyperandrogenism. This discrepancy reflects the fact that the patients were at different reproductive stages. Surgical removal of the ovarian mass resulted in clinical improvements in both cases. To the best of our knowledges, our patient is second case in which OART was seen in a woman without CAH.
The mechanism of ART formation still has not yet been fully elucidated. Kim et al. [4] mentioned that these tumors may arise from ectopic migration of residual adrenal cortex cells into the developing gonads because of the connection between the adrenal glands and gonads during embryonic development. Claahsen-van der Grinten et al. [5] suggested that defective primary sex cord regression, including all aberrant adrenal cells, during gonad development may be a causative factor in OART development.
In the literature, it has been shown that OARTs were related with distinct enzyme deficiencies associated with CAH, such as 21-hydroxylase, 11-hydroxylase, and 3-β hydroxysteroid dehydrogenase [6]. In our case, beside the negative results of Synacthen and genetic (CYP21A2 gene) tests, improvement of the patient’s clinical complaints (menstrual disturbances and hyperandrogenism signs, even including infertility) after the operation, supported exclusion of the diagnosis of CAH.
The diagnosis of OART masses by conventional imaging may be difficult because of the rarity of OART and sometimes small size of these tumors. Chen et al. [7] recommended examination of the ovaries and excision of any suspicious OART masses in female CAH patients who respond poorly to hormone therapy. In our case, we also suspected OART because the patient was unresponsive to hormone therapy and because of the patient’s ongoing complaints; additionally, the patient stated that this ovarian mass had been present for a long time, but it was not considered important since it was small.
Testicular adrenal rest tumors may be an important cause of infertility in men, as they lead to obstruction of the seminiferous tubules, azoospermia and gonadal dysfunction [8]. Disruption in the hypothalamic-pituitary-ovarian axis due to excessive androgens and excessive progesterone secretion in OART patients can causes infertility. Androgens can also directly inhibit folliculogenesis by having a negative effect on aromatase activity in granulosa cells. Additionally, progesterone hypersecretion in OARTs can affect the quality of cervical mucus and sperm penetration, accelerate endometrial maturation, reduce endometrial receptivity, decrease implantation, and exert negative effects on oocyte quality [9].
To the best of our knowledge, this is the first known case of OART causing infertility. There are three principal aspects that should be noted: first, this was an extremely rare case; second, the patient’s infertility and other hyperandrogenism-related complaints were successfully treated; and third, it was possible to exclude the diagnosis of CAH, which requires excessive steroid use and repeated treatments for infertility. Increased levels of androgen hormones by OARTs may lead to infertility by damaging the hypothalamo-hypophyseal axis. The appropriate diagnosis and treatment of these cases can prevent both excessive steroid use and recurrent treatments for infertility. The patient described herein conceived spontaneously following the excision of an OART.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Author contributions
Conceptualization: SK. Data curation: GO, HU. Formal analysis: HU. Funding acquisition: SK. Methodology: GO. Project administration: SK. Visualization: HU. Writing--original draft: HU. Writing--review & editing: HU.