Endoplasmic reticulum stress in periimplantation embryos
Article information
Abstract
Stress coping mechanisms are critical to minimize or overcome damage caused by ever changing environmental conditions. They are designed to promote cell survival. The unfolded protein response (UPR) pathway is mobilized in response to the accumulation of unfolded proteins, ultimately in order to regain endoplasmic reticulum (ER) homeostasis. Various elements of coping responses to ER stress including Perk, Ask1, Bip, Chop, Gadd34, Ire1, Atf4, Atf6, and Xbp1 have been identified and were found to be inducible in oocytes and preimplantation embryos, suggesting that, as a normal part of the cellular adaptive mechanism, these coping responses, including the UPR, play a pivotal role in the development of preimplantation embryos. As such, the UPR-associated molecules and pathways may become useful markers for the potential diagnosis of stress conditions for preimplantation embryos. After implantation, ER stress-induced coping responses become physiologically important for a normal decidual response, placentation, and early organogenesis. Attenuation of ER stress coping responses by tauroursodeoxycholate and salubrinal was effective for prevention of cell death of cultured embryos. Further elucidation of new and relevant ER stress coping responses in periimplantation embryos might contribute to a comprehensive understanding of the regulation of normal development of embryonic development and potentiation of embryonic development in vitro.
Introduction
There is increasing evidence linking endoplasmic reticulum (ER) stress coping responses with the development of diseases [1]. This is not surprising, given that the ER is a multifunctional essential organelle in eukaryotic cells; it functions as the critical entrance to the secretory pathways, and is also a regulator of Ca2+ homeostasis, as well as protein and lipid synthesis, all of which are highly sensitive to a variety of environmental, physiological, and pathophysiological conditions [1,2,3,4]. The ER is also closely associated with all cellular organelles, and therefore, capable of driving sophisticated mechanisms of intracellular signaling, including activation of transcriptional processes [2,3,5,6], Ca2+ mobilization [7], and influencing energy metabolism [8]. In this review, we briefly summarize the ER stress coping responses, particularly the unfolded protein response (UPR), and discuss the potential role of UPR as a physiologically relevant pathway in supporting and controlling proper embryonic development.
ER stress and UPR signaling
ER stress is caused by a disruption in cellular energy and/or nutrient homeostasis. It impairs ER functions such as protein folding and the heat shock response, and leads to altered Ca2+ levels and nutrient starvation. Activation of ER stress can result in the buildup of misfolded proteins in the ER and activation of the UPR as a coping strategy (Figure 1) [1]. The UPR involves distinct components designed to reestablish the protein synthesis machinery (Figure 1). These include (1) translational attenuation to arrest the entry of new proteins into the ER; (2) transcriptional activation of genes encoding proteins involved in protein folding to assist the maturation of proteins; (3) transcriptional activation of genes for components of the ER-associated protein degradation (ERAD) system to decrease the number of misfolded proteins; and if these are not successful, (4) activation of apoptotic pathways to eliminate defective cells [4,9,10]. Three ER transmembrane proteins: dsRNA-activated protein kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1), in combination with the ER molecular chaperone immunoglobulin binding protein (BiP, also known as glucose-regulated protein [GRP] 78), compose the UPR, an ER stress coping response. GRP78/BiP is a centrally located modulator/sensor of the UPR coping response. It is a monomeric, globular protein that functionally sorts and releases terminally misfolded substrates to the ERAD pathway. Under non-stressed conditions GRP78/BiP interacts with IRE, PERK, and ATF6, but upon an increase in misfolded proteins, GRP78/BiP is sequestered away from these inducers, leading to the activation of the UPR.
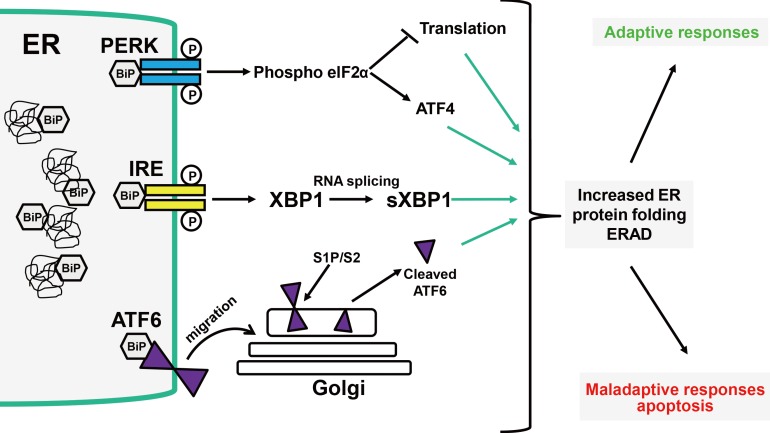
Unfolded protein response (UPR) pathways. Three endoplasmic reticulum (ER) transmembrane proteins--the ER kinase dsRNA-activated protein kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1), in combination with the ER molecular chaperone BiP, comprise the UPR reaction to ER stress. Under unstressed conditions, BiP directly interacts with IRE, PERK, and ATF6, but upon an increase in misfolded protein, BiP is sequestered away from these inducers, allowing activation of the UPR. PERK triggers its dimerization and autophosphorylation, followed by phosphorylation of eIF2α, preventing initiation of translation. ATF6 is an ER transmembrane protein cleaved in the Golgi. Cleavage of ATF6 produces a soluble basic leucine zipper (bZIP) transcription factor (cleaved ATF6) that binds to ER stress response elements (ERSE-I and II) to induce transcriptional activation of ER stress response genes. The transmembrane IRE1 dimerizes, leading to activation of kinase and endoribonuclease activity and splicing of the X-box binding protein 1 (XBP1) mRNA to produce an altered reading frame XBP1s mRNA. The XBP1s splice variant binds to the specific promoter elements, ERSE and unfolded protein response (UPRE), triggering transactivation of downstream UPR responsive genes, which are part of protein folding quality control and degradation machinery. ERAD, ER-associated degradation; sXBP1, splices XBP1.
Release of GRP78/BiP from PERK triggers its dimerization and autophosphorylation, followed by phosphorylation of eIF2α, at Ser51, which inhibits protein synthesis by sequestering the tRNAmet responsible for initiating the translation of nascent protein (Figure 1) [11]. During this period, only select mRNAs such as ATF4 are translated [12]. ATF4 transcriptional activity induces both pro-survival (early) and pro-apoptotic (late) transcriptional programs.
ATF6 is an ER transmembrane protein cleaved in the Golgi. Under non-stress conditions, ATF6 stays in the ER-associated GRP78/BiP. When BiP is sequestered away from ATF6, the ATF6 is translocated to the Golgi, where it undergoes cleavage by site-1 and site-2 proteases (S1P and S2P) (Figure 1) [13]. Cleavage of ATF6 produces a soluble basic leucine zipper (bZIP) transcription factor (cleaved ATF6) that binds to ER stress response elements (ERSE-I and II) to induce transcriptional activation of ER stress response genes. ATF6 can induce the expression UPR-induced genes including GRP78/BiP, proteins involved in ERAD, and XBP1 mRNA. Interestingly, ER stress triggered by Ca2+ depletion induces the formation of a nascent, partially glycosylated form of ATF6 with reduced interaction with calreticulin, and a faster rate of traversing the Golgi, resulting in higher transactivation of ATF6 gene targets [14].
Upon release of GRP78/BiP under stress conditions, IRE1 homodimerizes, causing a conformational change that is transmitted across the membrane, leading to activation of its cytoplasmic kinase activity, autophosphorylation, and activation of its endoribonuclease activity (Figure 1) [15]. IRE1 endoribonuclease activity cleaves 28S rRNA and specific microRNAs, inhibits protein synthesis, and also splices XBP1 (sXBP1) mRNA [16]. The protein product from the sXBP1 variant binds to the specific promoter elements, ERSE, and UPRE, triggering transactivation UPR responsive genes such as GRP78/BiP, calreticulin, and others, which are part of the protein folding quality control and degradation machinery. One part of the cellular recovery system that is turned on by the UPR is ER-associated degradation (ERAD) [5,17].
There is tight regulation of the three arms of the UPR with respect to the timing and response amplitude. The activation of each arm of the UPR is specific to the source of ER stress and governs cell fate, supporting either an adaptive response (cell survival) or a maladaptive response (cell death) [18]. Under prolonged and irreversible ER stress, cells become damaged and eliminated by apoptosis [19]. Under some ER stress conditions, UPR activates autophagy, a controlled self-degradation process that can promote cell survival by eliminating damaged cellular components [4]. IRE1 signaling could also engage apoptosis and autophagy [20,21,22,23]. The intensity, timing, and duration of the UPR appear to be important in determining cell fate. These observations suggest that ER stress coping responses may also play an important role in the maintenance of physiological events associated with organ function and/or development.
Targeting UPR, relief of ER stress
Although there is emerging evidence that activation of ER stress coping responses, including UPR, may play an important role in diverse human diseases [1,24,25,26,27,28], it is conceivable that these coping responses may also be critical under normal physiological conditions and during development. This may make UPR an attractive pathway to target for drug discovery [24]. Interestingly, there exists a group of small molecules that may stabilize protein folding, prevent protein aggregation, and consequently affect the severity of UPR activation. The most studied of these molecules are synthetic-fatty acid derivative 4-phenylbutyric acid and the natural bile acid tauroursodeoxycholic acid (TUDCA) [24,29]. Although the precise mechanisms of their action are not clear at present, these compounds have a profound impact on many pathologies including obesity pathways, brain ischemic-reperfusion, photoreceptor pathology, and steatosis, to name a few [24,30,31]. These findings suggest that targeting ER physiology and ER stress signaling and stress-induced coping responses has therapeutic potential for treating diseases related to ER stress [24].
ER stress is intrinsic as well as inducible in oocytes and preimplantation embryos
The development of preimplantation embryos is under the influence of various hormones and growth factors that originate from maternal tissues and/or the embryo proper. Preimplantation embryos synthesize and secrete a wide range of hormones and growth factors that promote embryonic survival; in the blastocyst stage, they increase in cell number via activation of transcription as well as protein synthesis [32,33,34,35]. During this synthetic activity, the embryos may experience an inherent level of ER stress [36] and consequently activate specific coping responses to recover homeostasis and support development or death. For example, in the mouse, the UPR contributes to preimplantation embryo death in the DDK syndrome [37], suggesting that the ER stress coping mechanism counters the stresses intrinsic to embryos. Mammalian early embryos placed in vitro are vulnerable to a variety of physicochemical stresses such as shearing, temperature changes, altered pH, and higher oxygen pressure, all of which are known inducers of ER stress coping responses. These stresses alter gene expression, epigenetic mechanisms, and metabolism, reducing the embryonic development and viability [38]. Although the influence of ER stress coping responses on cellular physiology in adult tissues has been widely studied, their effects on the oocytes and early embryos of mammals remains unclear.
Key regulators of the ER stress pathway in oocytes and preimplantation embryos
In a porcine model, the functional abundance of sXBP1 is low in mature oocytes and at the 1-, 2-, and 8-cell stages of embryos, but it is highly abundant at the germinal vesicle, 4-cell, morula, and blastocyst stages [39]. In mouse cumulus-oocyte complexes, fatty acid-induced ER stress impairs protein secretion, mitochondrial membrane potential, and embryo development. ER stress is a key mechanism mediating the developmental potential of oocytes [40]. In mouse oocytes, the cancer drug doxorubicin affects the mitochondria, PERK, and ER-associated caspase-12, while it inactivates PARP, resulting in the activation of apoptotic signals. This suggests that activation of ER stress coping responses may contribute, at least in part, to ovarian failure in women treated for cancer [41]. In mouse preimplantation embryos, ER stress-induced coping response molecules such as Perk, Ask1, Bip, Chop, Gadd34, Ire1, Atf4, Atf6, and Xbp1 are expressed [36,37,39,42,43]. Taking into account that PERK, IRE1, and ATF6 are the three major transducers of the UPR, the expression of the major transducers of the UPR suggests that preimplantation embryos use the UPR coping response as one mechanism to deal with preimplantation stress. In mice lacking UPR-associated genes such as GRP78 and Ppp1r15a/Ppp1r15b, the embryos do not survive beyond preimplantation development, suggesting the indispensable role of these ER chaperones and ER stress-induced coping responses in preimplantation embryos [42,44]. XBP1 is an important transcription factor and regulator of a subset of genes activated during ER stress-induced UPR. In Drosophila, Xenopus, and mice, XBP1 is essential for early embryonic development [45,46,47,48,49]. In mice, functional sXBP1 is abundant in the germinal vesicle of immature oocytes and on spindles in MII oocytes but is solely detected in the cytoplasm at the one-cell stage when sXbp1 mRNA is scarce. Nuclear sXBP1 abundance is high in preimplantation embryos [50]. This indicates that sXBP1 acts as an important regulator of ER stress-induced coping responses during specific developmental stages from oocyte to blastocyst. In mouse embryos, UPR inducers such as tunicamycin and sorbitol increased nuclear sXBP1 at the one- and two-cell stages and activate Xbp1 mRNA splicing at the 8-cell, morula, and blastocyst stages [36,50], indicating that the IREα arm of the UPR is activated as an important coping response and adaptation to ER stress in preimplantation embryos. In early mouse embryos, GRP78/BiP (HSPA5), a central regulator for the ER stress-induced UPR coping response, is essential for cell proliferation and protecting the inner cell mass from apoptosis [42]. Considering the importance of stress-activated protein kinase- mitogen-activated protein kinase kinase (SAPK-MAP3K) activation during cellular stress and the early stage of ER stress, as well as the indispensable role of stress-activated proteins and MAP3K for preimplantation embryonic development [51], ER stress manifested by SAPK-MAP3K is a normal event critical during preimplantation development [52]. The coupling of autophagy and UPR has been implicated in a variety of biological processes. Induction of autophagy promotes preattachment development of in vitro-produced bovine embryos by reducing ER stress, suggesting that autophagy/ER stress balance is important for the developmental competence of early embryos [53].
Effect of UPR inhibitors in oocytes and preimplantation embryos
The inhibition of ER stress-induced UPR responses by TUDCA was found to enhance the maturation and developmental potential of porcine oocytes by preventing ER stress-mediated apoptosis in vitro [39]. In mouse cumulus-oocyte complexes, fatty acid-induced ER stress coping responses are normalized by the UPR inhibitor salubrinal, demonstrating that ER stress is a key mechanism affecting oocyte developmental potential [40]. Preimplantation embryos, under in vitro conditions, experience an increase in reactive oxygen species (ROS), oxidative stress, heat-shock stress, and/or the culture stress due to insufficiency of maternally-derived embryotropic factors. Ultimately, this leads to the poor development and implantation failure of embryos [38]. Growth factors and ROS scavengers have been successfully used as a medium supplement in assisted reproductive technology (ART) programs to prevent poor outcomes for embryo survival under in vitro conditions [33,34]. Recently, TUDCA has also been used in the culture of preimplantation embryos [37,52,54]. TUDCA attenuates XBP1 mRNA splicing in embryos at the two-cell stage and abolishes DNA fragmentation induced by tunicamycin or sorbitol in blastocysts and recovered nuclear localization of the sXBP1 protein. In addition, TUDCA inhibits hyperosmolar-induced ER stress coping responses as well as the ER stress-induced apoptotic coping response during preimplantation embryo development [50,55].
ER stress signaling in post-implantation embryos
ER stress-induced coping responses may also play a critical role for embryo development after implantation. Indeed, increased ER stress in decidual tissue in pregnancy has been shown to be implicated in fetal growth restriction with and without pre-eclampsia [56]. Sustained ER stress acts as a cofactor of oxidative stress in decidual cells from patients with early pregnancy loss [57]. In decidual cells, excessive oxidative stress influences UPR pathways to activate ERAD by decreasing valosin-containing protein, which is a type II ER-associated protein and a member of the AAA±ATPase family that facilitates delivery of misfolded proteins to the proteasome, which results in cell damage, inhibition of cell growth, and activation of apoptosis [58]. Among UPR constituents, p-eIF2α, GRP94, and C/EBP homologous protein (CHOP) are increased in the placenta from intrauterine growth restriction and pre-eclampsia, indicating that ER stress responses are also inducible under diseased conditions [59,60]. Inositol-requiring enzyme 1α (IRE1α) or XBP1 null mice are unable to produce functional placentas, which is lethal to the embryo, indicating that the IRE1α arm of the UPR coping response is essential for placental development and embryonic viability [61]. In mice, cadmium-induced teratogenicity is associated with ROS-mediated induction of ER stress coping responses in the placenta [62]. Maternal exposure to cadmium resulted in activation of PERK, phosphorylation of placental eIF2α, and an increase in CHOP, indicating that UPR signaling is activated in the placenta due to cadmium-induced toxicity [62]. This further supports the notion that ER stress-induced coping responses are important mechanisms in embryos and extraembryonic tissues following exposure to environmental toxins. Furthermore, ER stress-induced coping responses in the maternal decidua and placenta may counteract developmental problems during implantation and postimplantation development.
Conclusions
Stress coping mechanisms are critical to minimizing or overcoming damage caused by ever changing environmental conditions. They are designed to promote cell survival. The UPR pathway is mobilized in response to the accumulation of unfolded proteins and to ultimately regain ER homeostasis. Various arms of the UPR, an ER stress coping response, are expressed in oocytes and preimplantation embryos, suggesting that ER stress as a normal physiological adaptive/coping mechanism plays a pivotal role in the development of preimplantation embryos. As such, UPR-associated molecules and pathways such as sXbp1 mRNA may become useful markers for the potential diagnosis of stress conditions for preimplantation embryos. Most importantly, UPR inhibitors, such as TUDCA, were found to effectively support the in vitro development of preimplantation embryos under stress conditions. Further identification of ER stress-induced coping responses in preimplantation embryos would be helpful for a comprehensive understanding of development from preimplantation through early postimplantation.
Acknowledgments
This paper was supported by grants from the Canadian Institutes of Health Research and the Brain Pool Program, the Korean Federation for Science and Technology (to MM) and by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A111441).
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.